<< Back to Resources Home Page
Epigenetics 101: Complete Guide to Understanding Epigenetics

Table of Contents:
-
What is Epigenetics?
-
History of Epigenetics
-
Types of Epigenetic Modifications
-
DNA Methylation & Methylation Variants
-
Histone Tail Modifications
-
Reading, Writing, and Erasing Histone Modifications
-
RNA Methylation & Other RNA Modifications
-
Non-Coding RNAs & Epigenetics
-
Applications of Epigenetics Research
-
Summary
Epigenetics is the study of how the environment, behavior, and other mechanisms cause changes to the genome that can contribute to the regulation of gene expression and other biological processes without changing to the underlying primary DNA sequences. These epigenetic mechanisms are mediated by chemical modifications of DNA, proteins (most commonly histones), and RNA. Epigenetic modifications are often reversible and can sometimes be heritable.
This article provides an introduction to the world of epigenetics, covers the history of this field in biology, discusses the factors and players that play a role in this process, and highlights which biological processes are influenced by epigenetic mechanisms.
What is Epigenetics?
While the precise definition of epigenetics has been a matter of debate since the term was first postulated by Conrad Hal Waddington in 1942, the word itself originates from the combination of epi-, the Greek prefix which means “over” or “upon” and genetics, the study of heredity. The combination implies something in addition to gene inheritance, hinting at a second layer of information on top of the primary information passed on. Most people now agree that epigenetics is the study of heritable differences in genetic expression passed down through successive generations of cells or organisms, without any change in the primary DNA sequence.
Although the study of epigenetics is still an emerging field, examples of its effects have been around us for millennia. Mule breeders unknowingly stumbled upon it thousands of years ago when they observed that a female horse crossed with a male donkey created a mule, while a male horse crossed with a female donkey created a hinny. A hinny is smaller and has stronger legs than a mule. Its temperament is also different than a mule, behaving more like a donkey (its mother) than a horse (its father). Both mules and hinnies are born from a cross between a horse and a donkey but depending on which genes are inherited from which parent, the traits are differently expressed.
A hallmark illustration of epigenetics is the transformation a butterfly makes inside a chrysalis from caterpillar to adult butterfly. Once the caterpillar has had enough to eat, it spins itself inside a cocoon and its body undergoes a metamorphosis from soft larvae to adult butterfly, sporting compound eyes, wings, and legs. Despite the morphology of these two states being completely different, the genes of the caterpillar and the adult butterfly don’t change. The only difference is how the genes are being expressed.
Our own bodies start out as a single cell zygote which eventually develops into an embryo and then a fully grown human with about 10 trillion cells. During the process, known as cellular differentiation, the zygote develops into pluripotent stem cells which can give rise to all of the different organs in the body. However, since all cells of the human body originate from this single-celled ancestor (except germ and immune cells), they have the same primary DNA sequence. Despite containing the same sequence, all the information is contained therein to form the different tissues and structures of the human body.
Because these processes can’t be explained solely by the primary DNA sequence of the genome there must be a second level of regulation at play. This is where the study of epigenetics comes in. It attempts to understand the mechanism by which the expression of particular genes is being modulated by some means other than the DNA sequence.
History of Epigenetics
It was Conrad Hal Waddington in 1942 (Waddington, 1942), who first coined the term “epigenotype,” stating that “between genotype and phenotype lies a whole complex of developmental processes.” This definition was built on the classical concept of “epigenesis,” which was introduced around 1650 by William Harvey describing development as a gradual process from the egg to a full-grown differentiated organism.
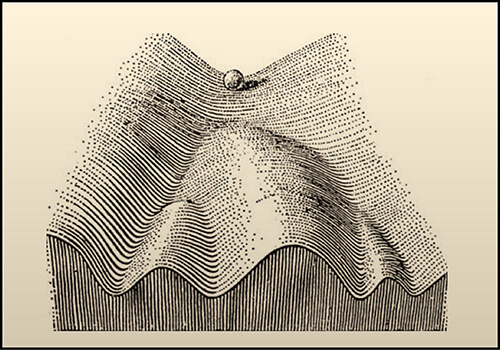
In 1957, after performing some experiments on embryos of D. melanogaster, Waddington proposed his model of the “epigenetic landscape.” The illustration of the model shows a ball rolling down an inclined surface containing branching mountains and valleys. Each time the ball comes to a crossroads there is a decision to make and the ball can take different paths. This picture is a metaphor for the decisions that need to be made in the development and differentiation of a cell. The underlying genes act to structure the “landscape,” whereas the presence or absence of genes determines which path shall be followed from a certain crossroads.
A year later, two cellular control systems were described by David Ledbetter Nanney. The first was a “library of specificities”, which corresponded to genes encoded in the primary sequence of the DNA. The second system, termed “auxiliatory mechanisms,” determined which set of features were expressed in any particular cell. In an effort to simplify, he proposed using the terms “genetic systems” and “epigenetic systems,” referencing Waddington’s paper and underscoring the dependence of the epigenetic processes on the genetic system.
Chromatin Uncovered
While it wasn’t until the turn of the millennium that the molecular mechanism of “epigenetics” received much focus, the groundwork was being laid for its understanding long before. In the late 19th century, Walther Flemming was staining and studying the structures of the cell nucleus both before and during cell division. This led to a landmark work in which he published what are likely the first images of human chromosomes.
It was he who first coined the word “chromatin,” describing the threads he saw in the nucleus that could easily be stained. “Achromatin” were those structures that didn’t stain. He did not understand exactly what these structures were, but knew it was something important. He wrote “... The word chromatin may stand until its chemical nature is known, and meanwhile stand for that substance in the cell nucleus which is readily stained.” The term “chromatin” still stands to this day, to describe the thread-like complex of DNA and protein found in cells.
Histones, the family of alkaline proteins that provide the packing structure for DNA in the nucleus, were discovered shortly after by Albrecht Kossel. In addition to his well known discovery of the organic nucleobases that make up nucleic acids, he reported the isolation of an acid-extractable peptone-like substance from the nuclei of goose red blood cells. It wasn’t clear at that time what its purpose was, but he suggested that it might be bound to nucleic acid, naming it “histone.”
The first reports that histones might be involved in epigenetics came in the 1960s. An association between the acetylation of histones and the regulation or RNA synthesis was reported in PNAS by Allfrey, Faulkner, and Mirsky in 1964. The paper presented data which suggested RNA synthesis was inhibited more by histones in their native state, than by acetylated histones. They suggested that acetylation of histones may be a means of “…switching-on or -off RNA synthesis.” The hypothesis that histones might regulate gene expression was a critical step in the future understanding of epigenetic mechanisms.
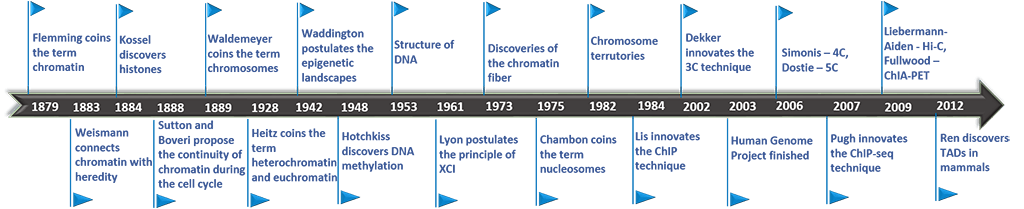
Not long after this, nucleosomes, the basic structural unit of chromosomes consisting of a length of DNA wound around a core of histones, were first visualized. In 1973, Ada and Don Olins successfully used electron microscopy to capture images of spherical particles in chromatin about 70 angstroms wide, which they called “ν bodies.” These were further observed to be connected by strands 15 angstroms wide, making them look like “beads on a string.” In 1975 Roger D. Kornberg went further, describing the basic structure of chromatin as “... a repeating unit of two each of the four main types of histones and about 200 base pairs of DNA. A chromatin fiber may consist of many such units forming a flexibly jointed chain.” It was that same year the “nucleosome” received its present name.
The discovery and definition of the nucleosome changed the perception of chromatin. It was now seen as being coiled on the outside of a globular histone core, which makes it accessible from the outside for the interaction and binding of nuclear proteins.
Shortly after, a race to solve the crystal structure of the nucleosome began. The first nucleosome structure was determined by Timothy Richmond in 1984 to 7.0 Å. And finally, in 1997, Karolin Luger and colleagues solved the crystal structure of the nucleosome to 2.8 Å, clearly showing how each histone octamer is assembled, with 146 base pairs of DNA organized into a “superhelix” around it. Multiple histone/histone and histone/DNA interactions were determined which paved the way for understanding how specific amino acids on histone tails contact and affect DNA.
Many of the important findings on chromatin were nicely summarized in a landmark review paper by Thomas Jenuwein and C. David Allis in 2001 called “Translating the Histone Code.” There, it was proposed that posttranslational modifications on histone tails may lead to synergistic or antagonistic interactions with chromatin-associated proteins. Furthermore, they proposed that the interplay and combination of these different marks could lead to distinct local environments which favored transcriptionally active (euchromatin) or transcriptionally silent (heterochromain) chromatin states. It was here that the remodeling of chromatin was proposed as a pivotal epigenetic regulatory mechanism, “with far-reaching consequences for cell fate decisions and both normal and pathological development.”
DNA Methylation Uncovered
Research on DNA methylation was being carried out in parallel to research on histones. Eukaryotic DNA methylation was first discovered in 1948 by Rollin Hotchkiss. By using paper chromatography, he found that a fraction of cytosines in calf thymus DNA were modified. He hypothesized that it was 5-methylcytosine (5-mC) because it separated from cytosine similarly to the way thymine (aka methyluracil) separated from uracil.
However, it took until the mid 1970s until the relationship between DNA methylation and epigenetics began to unfold. In 1975, AD Riggs published a model tying X-chromosome inactivation to sequence-specific DNA methylases. X-chromosome inactivation occurs because females receive two copies of the X-chromosome while males only receive one copy. To prevent female cells from producing twice as many X-linked gene products as males, one of the chromosomes must be silenced. Which one is inactivated is random for each cell, but it remains inactivated for the lifetime of the cell. The inactivated X chromosome is kept from expressing its genes in part through high levels of DNA methylation.
In the same year, R. Holliday and J.E. Pugh proposed a possible mechanism by which DNA modification enzymes in eukaryotes might control gene activity during development, based on known features of DNA modification in bacteria. While there was not yet direct evidence of specific DNA modification enzymes in eukaryotes, they made an argument for why these should be experimentally searched for. First, it was known that methylation of DNA was not random; secondly methylated CpGs were observed frequently and yet were located less commonly in the DNA than by chance; and thirdly, methylases had been identified in sea urchin embryos. All of this pointed to a likely mechanism where DNA methylation was actively controlled in the cell.
It was Adrian Bird who pioneered the characterization of DNA Methylation at CpG Islands in the 1980s and 1990s. Identification of methylated genetic regions in vertebrates was made possible by using their sensitivity to a methyl-sensitive restriction enzyme. It was discovered that methylated regions occur as discrete 'islands,' usually 1-2 kbp long, that are dispersed in the genome about one every 100 kbp. Those Islands later became famous under the term CpG Islands. Furthermore, he found that CpG islands are usually found either completely methylated or completely unmethylated within or near the promoter regions of genes, suggesting they were involved in gene regulation and transcription.
In the years following, there has been much focus on determining endogenous patterns of DNA methylation and how these patterns may be passed down through successive generations of cells or organisms. A number of different enzymes have been found to be responsible for both de novo methylation and for maintenance of previously methylated sites.
Types of Epigenetic Modifications
The organization of genetic material in the nucleus has profound effects on all processes that require access to DNA. Chromatin is a dynamic structure. Covalent modifications made to either the proteins or DNA that make up chromatin can alter its conformation. However, the two most studied of these are covalent modifications of DNA at cytosines and the post-translational modification (PTM) of histone tails at lysines, arginines, serines, and threonines.
These types of modifications can change how proteins and DNA interact with each other, changing the physical state of the chromatin. Some modifications result in loosely packed chromatin, known as euchromatin, making it possible for transcription factors to bind to promoters upstream of genes and allowing RNA polymerase II to bind to DNA. Euchromatin makes up the most active regions of the genome. In contrast, other modifications create tightly packed chromatin, known as heterochromatin, where accessibility to transcription proteins is limited, create inactive regions of the genome.
The covalent modifications of both DNA and histones are carefully controlled by an array of proteins. Some are involved in adding modifications. Others remove them. In this fashion, gene expression can change and respond as necessary, either to environmental changes or mitosis. Furthermore, these modifications can be passed on to daughter cells after mitosis or even selectively to future generations through the germline, without changing the primary DNA code.
Histone H3K4 Methylation – The Promoter Mark
Histone H3K4 is most well-known as a site for trimethylation, marking active promoters, and for this reason we refer to this as the promoter mark. However, H3K4 can also be acetylated, just upstream of the trimethylation.
H3K4me3 (trimethylation of Histone H3 on Lysine 4) is present at the transcriptional start site (TSS) of active promoters and is written by the WDR5 histone methyltransferase enzyme. KDM5B/JARID1B is a histone demethylase enzyme that can reverse this mark.
H3K4me1 (monomethylation of Histone H3 on Lysine 4) is a mark that is present on poised and active enhancers. It is often measured along with H3K27ac, where the presence of both indicates an active enhancer but presence of only H3K4me1 indicates a poised enhancer. The writer enzymes are KMT2C and KMT2B (MLL3 and MLL4, respectively). The lysine demethylase KDM1A/LSD1 act as an eraser of this mark.
H3K4ac is found on promoters of active genes, just upstream of the TSS, which is usually marked by trimethylation.
Histone H3K9 Methylation – The Heterochromatin Mark
Histone H3K9 is most well-known as a site of di- and trimethylation, maintaining closed chromatin, and for this reason we refer to this as the heterochromatin mark. However, H3K9 can also be acetylated, marking promoters of active genes.
H3K9me2 and H3K9me3 (dimethylation and trimethylation of Histone H3 on Lysine 9 H3K9me2 and H3K9me3) are marks found on heterochromatin. These marks are crucial in maintaining cell lineage commitment during differentiation. They restrict certain parts of the genome from being transcribed and represent a barrier against cellular reprogramming. The writers of these marks are SET-domain containing methyltransferases SETDB1, SUV39H1, and SUV39H2. A major reader of these marks is Heterochromatin Protein 1 (HP1). Once bound to H3K9me2 and H3K9me3, it can recruit more repressive histone modifiers. However, in stem cells, where pluripotency is required, the H3K9me2 and H3K9me3 marks are erased by the histone demethylase proteins KDM3A and KDM4C, respectively, from loci required for stem cell renewal.
H3K9ac is found on promoters of active genes, whereas H3K9me1: is found at the transcriptional start sites (TSS) of active genes.
Histone H3K27 Methylation & Acetylation – The Dual-Purpose Histone Tail Residue
Post-translational modification of Lysine 27 on Histone H3 is currently the most reliable histone mark indicator of whether a regulatory element is on or off. Briefly, acetylation indicates open chromatin and trimethylation indicates closed chromatin.
H3K27me3 (trimethylation of Histone H3 on Lysine 27) marks heterochromatin at repressed enhancers and promoters. The most common writer of this mark is EZH2 and erasers include the demethylating enzymes JMJD3 and UTX. Readers of the H3K27me3 mark are the polycomb complexes PRC1 and PRC2. More specifically, PRC2 initiates this silencing and PRC1 acts to maintain the closed chromatin memory state, required after differentiation. The eraser of this mark is the lysine demethylase KDM6A.
H3K27ac (acetylation of Histone H3 on Lysine 27) is perhaps the most widely-measured and highly-abundant mark of activation and open chromatin. Acetylated H3K27 is present at active promoters and active enhancers. The histone acetyl transferase proteins p300 and CBP are the main writers of H3K27ac and the erasers are HDAC1 and HDAC2.
Histone H3K36 Methylation – The Active Transcription and DNA Replication/Recombination/Repair Mark
Methylation on H3K36 appears at sites of active transcription, repair of DNA damage, replication, and recombination. The SET domain proteins are the writers of mono-, di-, and trimethylation at this residue. Readers of H3K36 methylation are proteins containing the plant homeodomain finger, as well as the chromo, Tudor, and PWWP domains. The KDM (histone lysine demethylase) family proteins are the erasers of this mark. Interestingly, H3G34 is mutated in several pediatric cancers and this mutation can block methylation of H3K36, thereby blocking DNA damage repair and other functions mediated by H3K36 methylation.
H3K36me2 (dimethylation of Histone H3 on Lysine 36) has a role in transcription initiation (deposited by SETD2 at transcriptional start sites). When SETD2 interacts with a phosphorylated RNA Pol II, it further methylates histone H3 up to H3K36me3 and elongation begins. H3K36 dimethylation also has a role in intergenic regions. H3K36me2 can be written by the histone methyltransferase NSD1 (also a SET domain protein) and this mark recruits the DNA methyltransferase DNMT3A for maintenance of DNA methylation. Haploinsufficiency of NSD1 (Sotos syndrome) results in less H3K36 dimethylation and deficiency of DNA methylation.
H3K36me3 (trimethylation of Histone H3 on Lysine 36), as described above, is deposited by the writer SETD2, after it binds to RNA Polymerase II, and this mark plays a role in transcription elongation. H3K36 methylation can be erased by the histone demethylase enzymes KDM2A-B and KDM4A-D.
H3K36me1 is less well-characterized but is known to be associated with active transcription of genes, much like the di- and trimethylation modifications of H3K36. A reader of this mark is the RPD3 histone deacetylase complex, which is recruited by H3K36me1 to become an eraser of acetylated lysine residues on all histone subunits.
H3K36ac has been characterized in plants as a modification found at the 5’ ends of actively transcribed genes.
DNA Methylation & Methylation Variants
DNA methylation is an important regulator of gene expression and genomic organization. It occurs at the cytosine bases of eukaryotic DNA, which are converted to 5-methylcytosine (5-mC) by DNA methyltransferase (DNMT) enzymes. DNA methylation appears almost exclusively in the context of CpG dinucleotides. These dinucleotides are relatively rare in the mammalian genome and tend to be clustered in what are called CpG islands. Approximately 60% of gene promoters are associated with CpG islands and are normally unmethylated, meaning they are transcriptionally active. CpG methylation of gene promoters is usually associated with transcriptional silencing.
DNA methylation is carried out in two distinct enzymatic processes: de novo DNA methylation and maintenance DNA methylation.
De novo DNA methylation involves the addition of methyl groups to previously unmethylated DNA and is mainly present in embryonic stem (ES) cells, early developing embryos, and developing male and female germ cells. It’s largely suppressed in adult differentiated somatic cells. De novo DNA methylation is carried out by DNA methyltransferases 3a and 3b (DNMT3A, DNMT3B) and is believed to play a critical role in the establishment of genomic imprinting, the differential expression of paternal and maternal alleles during gametogenesis and in the offspring.
Maintenance DNA methylation is meant to preserve DNA methylation after every cellular DNA replication cycle. It’s primarily carried out by DNA methyltransferase 1 (DNMT1) and occurs during DNA replication in adult cells on hemi-methylated DNA (DNA with only one methylated strand). Hemi-methylated DNA is created during the course of DNA replication, when unlike the parent strand, the newly synthesized strand is not methylated. DNMT1 binds to these hemi-methylated CpG sites and methylates the cytosine on the newly synthesized stand. This maintains established CpG methylation patterns through mitosis. Otherwise, DNA methylation would become diluted over many cell divisions and ultimately be lost.
DNA methylation can also be reversed. TET enzymes, a family of ten-eleven translocation (TET) methylcytosine dioxygenases are central for DNA demethylation. These enzymes use 5-mC as a substrate converting it to 5-hmC. Further action by TET can catalyze the oxidation of 5-hmC to 5-formylctyosine (5-fC) and then to 5-carboxyctyosine (5-caC). Both 5-fC and 5-caC can be removed from the DNA sequence by base excision repair and replaced by an unmethylated cytosine.
While certain enzymes are involved with the writing and re-writing of the DNA methylation code, other proteins are used for reading this epigenetic mark. Methyl-CpG-binding domain (MBD) proteins are highly specific for binding DNA that contains one or more symmetrically methylated CpGs. Each MBD can cover up approximately 12 nucleotides of DNA around a methyl CpG pair. The MBD protein acts as a structural protein, recruiting a variety of histone deacetylase (HDAC) complexes and chromatin remodeling factors, which lead to chromatin compaction and subsequent transcriptional repression.
DNA methylation is important for a number of cellular functions, such as embryonic development, genetic imprinting, X chromosome inactivation and control of gene expression. Alterations in normal DNA methylation patterns have been shown to correlate with a wide variety of disease processes such as cancer, congenital disorders, and cardiovascular disease. In cancer, hypermethylation of promoter CpG islands can cause loss of expression of critical genes which keep cell division in check. Methylated cytosines are more likely to become mutated in the genome than unmethylated cytosines as they are more likely to undergo spontaneous deamination. Deamination of cytosine converts it to a thymine. The result is a mismatched GT pair which must then be repaired. If not repaired before replication a permanent C to T mutation can be written into the genome.
Mutations or altered expression in the proteins which alter and read DNA methylation, such as DNMTs, TETs, and MBDs have also been implicated in a number of diseases. Mutations in the methyl CpG binding protein 2 (MECP2) causes Rett syndrome. Mutations in both the DNMT3A and TET2 proteins are associated with development of cancer.
DNA methylation has been shown to be strongly tied to the aging process as well. During aging, DNA methylation patterns change in tissue and cells in a predictable pattern. In 2018 a Nature paper by Steve Horvath and Kenneth Raj describe the successful use of DNA methylation data for biological aging of tissue, giving rise to a sort of “epigenetic clock.” A better understanding of normal DNA methylation patterns, together with new tools to assay it in cells, could lead to future diagnostics for and treatments for DNA methylation related conditions.
Histone Tail Modifications
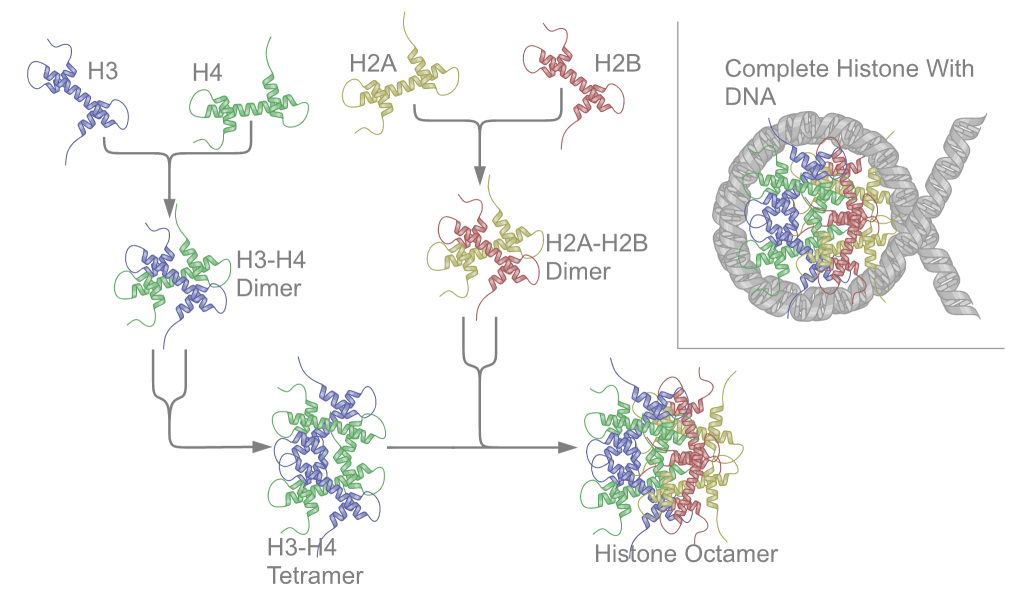
Nucleosomes are the basic unit of chromatin and consist of a 147 bp segment of DNA wrapped around a disc of eight histone proteins. This histone octamer core is formed by histone proteins H2A, H2B, H3 and H4. The ~18 kDa histone proteins are evolutionary well conserved and feature a “helix turn helix turn helix” motif. To form the nucleosome, H3 and H4 form a heterodimer which then dimerizes into a H3-H4 tetramer. This eventually forms a histone octamer with two H2A-H2B heterodimers.
Histones contain many positively charged amino acid residues, which form favorable electrostatic interactions with the negatively charged phosphate groups in the DNA backbone. In this way DNA wraps tightly around the histone-octamer core. A number of these positively charged residues reside on the N-terminal tail of each histone, which protrudes from the core of the octamer and is accessible from the outside of the nucleosome. These tails play roles in key inter- and intra-nucleosome interactions.
Modification of histone tails mainly takes place at lysine residues; but also takes place at some arginine, threonine, and serine residues. Lysine and arginine residues can be either methylated or acetylated. Serine and threonine residues can be phosphorylated.
Acetylation of a lysine residue results in the loss of a positive charge. This weakens its association to the DNA, causing a more open chromatin structure. Histone acetylation has been implicated in a number of cellular processes, but its most common function is in transcriptional activation. Histone acetylation is heavily targeted to gene promoter regions. By opening up the chromatin structure in these regions, transcription factors gain access and increase gene expression.
The effects of lysine methylation are a bit trickier to predict. It depends both on which lysine on the tail is methylated and whether it’s mono-, di-, or tri-methylated. The result can be either transcriptional silencing or activation. Methylation events that weaken chemical attractions between histone tails and DNA will increase transcription just as acetylation does, because they enable the DNA to uncoil from nucleosomes so that transcription factor proteins and RNA polymerase can access the DNA. Methylation of lysine and arginine residues function as a major determinant for formation of transcriptionally active and inactive regions of chromatin and is crucial for proper programming of the genome during development.
Phosphorylation of serine and threonine residues is less understood but has been shown to be particularly important for DNA repair and mitosis. For example, phosphorylation of serine 10 and serine 28 on the tail of histone H3 occurs early in mitosis when chromosome condensation is induced during S-phase.
Modifications in the histone tails of all four core histones, H2A, H2B, H3 and H4 are involved in gene regulation. However, tails of histones H3 and histone H4 have been shown to be especially important. The histone H3 N-terminal tail contains an arginine at position 2, nine lysine residues at positions 4, 9, 14, 18, 23, 27, 36, 56 and 79, and a serine at position 28. Histone H4 contains an arginine at position 3, five lysine residues at positions 5, 8, 12, 16, and 20, and a serine at position 1.
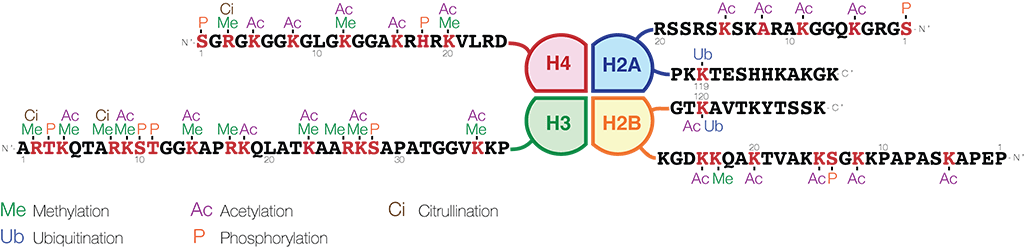
Image credit: https://en.wikipedia.org/wiki/Histone - CC BY-SA 4.0
Reading, Writing, and Erasing Histone Modifications
The possibilities of modified histone residues in various combinations may seem endless. However, like with DNA methylation, there are regulatory mechanisms in place which are responsible for the maintenance or change of the adequate chromatin environment in each cell or at specific genes. The special proteins involved with this task are known as histone readers, writers, and erasers.
Readers of Histone Modifications
Readers of histone PTMs are enzymes that bind to the modifications at histone tails and process this information. Most of such proteins contain either a bromodomain, to bind to acetylated amino acid residues, or a chromodomain, to bind to methylated amino acid residues.
After binding, they recruit other factors like chromatin-remodeling complexes to change the local chromatin environment. One example of a bromodomain containing reader is the SWItch/Sucrose Non-Fermentable (SWI/SNF) complex. It’s known as a “access remodeler” as it is able to alter the position of nucleosomes along the DNA by destabilizing histone-DNA interactions. It promotes gene expression by exposing binding sites so that transcription factors can bind more easily. The SWI/SNF subunits are often absent in cancer cell lines.
An example of a chromodomain containing reader is the chromodomain-helicase-DNA-binding protein 4 (CDH4). It is part of a both a deacetylase and nucleosome remodeling complex which acts to shut down transcription. An autoimmune condition known as dermatomyositis is caused by antibodies against this protein.
Writers of Histone Modifications
Writers of histone PTMs are chromatin-modifying enzymes that actively modify amino acid residues on the tails of the core histones. These enzymes include histone acetyltransferases (HATs), histone methyltransferases (HMTs), and phosphorylases.
One important member of the family of HATs is p300, which regulates cellular growth and differentiation. The p300 enzyme can acetylate all four core histones and regulates transcription via chromatin remodeling. Furthermore, it binds to transcription factors and functions as a coactivator of transcription. P300 is important in preventing tumor growth which makes it an interesting target for biomedical research.
An important member of the family of HMTs is the enhancer of zeste homolog 2 (EZH2), which functions as a mediator of epigenetic transcriptional silencing. It catalyzes the mono-, di- and tri-methylation of Histone H3Lys27, leading to the formation of heterochromatin and the silencing of gene function. The FDA recently approved an inhibitor of EZH2, called Tazverik, by Epizyme Therapeutics for the treatment of certain cancers.
Erasers of Histone Modifications
Erasers of histone PTMs remove modifications from the respective amino acid residues. These enzymes include histone deacetylases (HDACs) and histone demethylases (HDMs).
Histone deacetylases remove acetyl groups from histone tails, increasing their positive charge and encouraging high-affinity binding between the histones and DNA backbone. The increased DNA binding condenses DNA structure, preventing transcription. The family of HDACs is currently divided into 5 classes, based on function and DNA sequence similarity. Members of class I, II and IV, known as the “classical” HDACs are inhibited by trichostatin A (TSA) and have a zinc-dependent active site. Class III enzymes are not affected by TSA and are all NAD+ dependent proteins. Inhibitors of HDACs have been used for many years in psychiatry as mood stabilizers and anti-epileptics. The most notable example is valproic acid.
Histone demethylases remove methyl groups from histone tails. For many years, it was believed that histone methylation was irreversible. However, in 2004, the histone demethylase LSD1 was discovered and since then many more histone demethylases have been found. These have been divided into the two families: KDM1 and the JMJC Domain-containing histone demethylates. Some of these catalyze the removal of mono- or di-methylated residues, while others catalyze the removal of tri-methylated residues. Like many other epigenetic proteins involved in transcription, altered expression of histone demethylases can result in aberrant histone modifications that can drive cancer progression, metastasis and resistance to therapy.
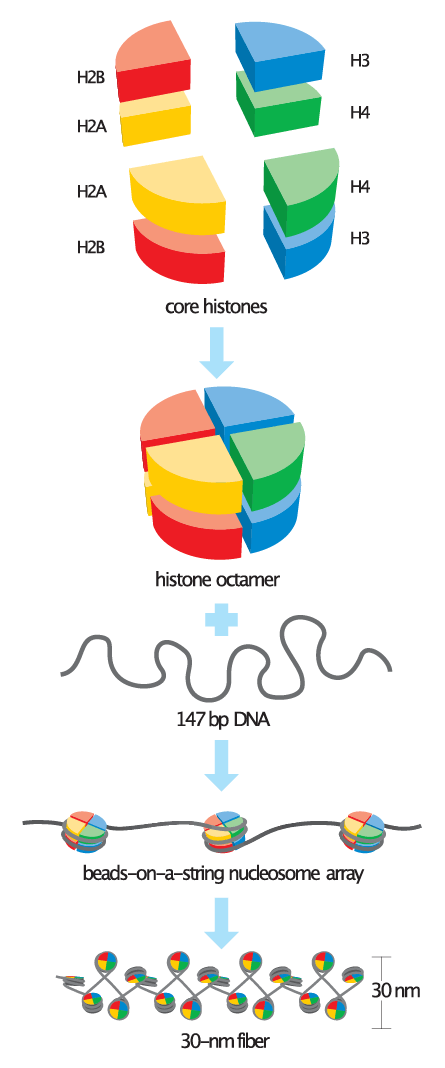
Image credit: Wikimedia Commons - Basic_units_of_chromatin_structure.svg - CC BY-SA 4.0
RNA Methylation & Other RNA Modifications
In addition to DNA methylation and histone PTM modifications, it has been more recently determined that RNA also contains post-transcriptional chemical modifications that are involved in epigenetics. Messenger RNA (mRNA), the molecule that carries genetic information from DNA to ribosomes, and long non-coding RNA (lncRNA), which are never translated into proteins can carry an N6-methyl-adenosine (m6A) modification, among others.
The formation of m6A residues in eukaryotic messenger RNA (mRNA) was first described in the 1970s; but it gained traction in 2011 when Guifan Jia et. al. found that knockout of fat mass and obesity-associated protein (FTO) let to an increase in the amount of m6A in mRNA, where overexpression of FTO resulted in decreased amounts of m6A.
The m6A modification is one of the most abundant RNA modifications in eukaryotes and occurs at the consensus sequence RR(m6A)CH. This base modification is present on average in ~3 sites of every mRNA in mammals and has been shown to be critical to cell differentiation, animal development, and a range of biological signaling and stress response. It is enriched in the 5′ UTR, in large exons, and in the proximity of the stop codon in the 3′ UTR of mRNAs.
Similar to the field of DNA methylation and histone PTMs, there are readers, writers, and erasers of RNA modification.
Readers of RNA methylation can be divided up into two families. Some, like members of the YTH family, directly read the presence of m6A. Others, like some hnRNPs, don’t read the mark itself, but read the secondary structure of the RNA, which can change upon modification. Functional characterizations of YTHDF2 have shown it affects cytoplasmic localization and mediates the decay of methylated mRNA. YTHDF1 promotes translation of methylated mRNA by facilitating translation initiation. Other readers affect mRNA storage, transport, and cellular localization.
Writers of m6A were first discovered in 1994. In mammalian cells, two very important writers of m6A are METTL3 (methyltransferase-like 3) and METTL14 (methyltransferase-like 14). These two proteins form a stable heterodimer core complex of METTL3–METTL14 that functions in cellular m6A deposition on mammalian nuclear RNAs. METTL3 is the catalytically active subunit that catalyzes m6A methylation, while METTL14 plays a structural role critical for substrate recognition.
As mentioned above, characterization of the protein FTO (fat mass and obesity-associated protein) first rekindled the interest in the m6A RNA Modification. FTO, an eraser protein, can demethylate the m6A in RNA molecules. Alterations in the gene have been observed to go along with increased body mass. Another RNA demethylase is ALKBH5. Just like FTO it oxidatively reverses m6A in mRNA in vitro and in vivo. When ALBKH5 is knocked out, the level of m6A in male mouse mRNA increases and spermatogenesis and fertility are impaired.
Besides m6A, a number of other modifications exist in mammalian mRNA, including N1-methyladenosine (m1A), 5-methylcytosine (m5C), pseudouridine, and 2′-O-methylation (2′OMe). This collection of chemical modifications modulates nearly all aspects of RNA metabolism and related physiological processes, adding another layer to the already complex gene expression regulation pathways in eukaryotes, particularly in mammals.
Non-Coding RNAs & Epigenetics
There is a whole family of RNAs that do not follow the central dogma of molecular biology and are not translated into proteins. These types of non-coding RNAs (ncRNAs) include transfer RNAs (tRNAs) and ribosomal RNAs (rRNAs), as well as small RNAs such as miRNAs, siRNAs, piRNAs, snoRNAs, snRNAs, exRNAs, scaRNAs, and lncRNAs. Many of these are involved in epigenetic regulation, including microRNAs, siRNAs, piRNAs, snoRNAs, and lncRNAs. Until the 1980s, little was known about the regulatory effects of RNAs in general, let alone ncRNAs. And then in the early 2000s, with the discovery of the miRNAs, the ncRNA revolution gained momentum and more members of the family were discovered.
MicroRNAs
MicroRNAs (miRNAs) are 22 nucleotide single-stranded RNAs that function in post-transcriptional RNA silencing pathways by base pairing with their target mRNA, leading to degradation or repression of translation. miRNAs originate from pri-miRNA molecules, RNA transcripts, approximately 70-80 nucleotides long that usually come from intronic regions of their target gene. Pri-miRNA fold back on themselves to form short hairpins and can be cut by an enzyme called DICER to yield mature miRNA. The mature miRNA can then become incorporated into the multi-protein complex known as the RNA-induced silencing complex (RISC). The miRNA acts as a template for mRNA target identification and RISC complex cuts the mRNA.
Small Interfering RNAs
Small interfering RNAs (siRNAs) are small (20-27 bp) double-stranded RNAs which also can become incorporated into RISC for mRNA target identification. siRNAs are formed when the DICER enzyme cuts long dsRNAs. Once part of RISC, the siRNA is unwound to form single stranded siRNA, with one strand, the guide strand remaining in the RISC-complex and the other strand, the passenger strand, becoming degraded. The RISC complex, now containing the guide strand, can bind and then cleave complementary mRNA.
miRNAs and siRNAs interact slightly differently with mRNA. miRNA does not need to have perfect complementarity to its target mRNA in order to bind and cause mRNA degradation in the RISC complex or translational repression. Only a short ~6 nt region known as the seed sequence needs to match. siRNA, on the other hand, must compliment perfectly with its mRNA target. Because of this miRNA has a much broader specificity of action and can bind to and regulate the expression of multiple mRNAs (>100) whereas an siRNA contains a perfect match with its mRNA target and so typically only affecting expression of one specific target mRNA.
Piwi-Interacting RNAs
Less is known about the other non-coding RNA molecules. Piwi-interacting RNAs (piRNAs) are single-stranded and between 21-31 nt. Their origin is still not fully understood, but they also interact with RISC. Given their similarity to other non-coding RNAs it is thought that they function in gene silencing. piRNAs are found in mammal testes and ovaries, and likely play a role in spermatogenesis and embryogenesis.
Small Nucleolar RNAs
Small nucleolar RNAs, snoRNAs, are located in the nucleolus and unlike the aforementioned ncRNA-species do not have such a defined size window with their length ranging from 60 to 300nt. Their primary function is to guide chemical RNA modifications to other RNA species like mRNAs, tRNAs, or rRNAs. SnoRNAs can be divided into two classes: C/D box snoRNAs and H/ACA box snoRNAs each being responsible for the delivery of different RNA modifications. C/D box snoRNAs form a stem-box structure and are associated with methylation whereas H/ACA box snoRNAs form a hairpin-hinge-hairpin-tail structure and are associated with pseudouridylation.
Long Non-Coding RNAs
Long non-coding RNAs, lncRNAs, are RNA transcripts of 200nt or larger which are not translated into proteins. LncRNAs are involved with epigenetic regulation with the most prominent example being X-inactivation. During the early stages of development one of the X chromosomes is silenced by being coated by multiple layers of inactive chromatin marks. This process starts with the expression of the Xist lncRNA and leads to a loss of H3K9 acetylation and an irreversible accumulation of H3K17 methylation, causing transcriptional shutdown of one of the X chromosomes.
Applications of Epigenetics Research
The importance of epigenetics on diseases like cancer, diabetes, obesity, and aging has become increasingly evident in recent years. Changes in DNA methylation, post-transcriptional histone modifications, and non-coding RNAs have been linked to numerous conditions.
Hypermethylation of promoter CpG islands in DNA repair genes and tumor suppressor genes results in a reduction in their gene expression. This hypermethylation has been found in a large fraction of cancers including bladder, stomach, thyroid, colorectal, brain, lung, prostate and breast cancer.
Histones are linked to tumorigenesis through dysregulation of PTMs modifications and their modifiers. In cancers, these alterations can result in the inappropriate activation of oncogenes or, conversely, the inappropriate inactivation of tumor suppressors.
Both miRNAs and lncRNAs have been shown to be involved in the pathophysiology of cancer, cardiovascular and neurological disorders. The result has been an ever-growing interest in using epigenetics to diagnose or treat conditions where these changes occur.
Perhaps the area of epigenetic clinical research that has garnered the most interest is in the analysis of liquid biopsies. A liquid biopsy is a non-invasive method to obtain biological samples from a patient. Liquid biopsies are most commonly blood, plasma, or serum samples, but they can also be derived from other biological liquids. Liquid biopsies can be analyzed for many different types of biomarkers and used in diagnostic or prognostic tests to monitor disease progress, responses to treatments, or to look for early warning signs of diseases.
Liquid biopsies are specifically useful to look for human cancers, as tumors shed both DNA and cells which end up in blood, urine, or saliva. The liquid biopsy samples are analyzed for the presence of circulating tumor cells (CTCs) or circulating cell-free DNA (cfDNA) containing changes associated with cancer. While initial liquid biopsy assays sought to look for genetic mutations associated with cancer, there is now much interest in using them to look for epigenetic changes in DNA methylation patterns, histones, nucleosomes, or miRNAs.
Summary
Epigenetics is a fundamental biological process that contributes to many cellular mechanisms such as regulation of gene expression, DNA replication and repair, and higher order nuclear organization. Epigenetic mechanisms involve modifications to DNA and chromatin-associated proteins, rather than changes to the primary DNA or amino acid sequences. Although the term “epigenetics” and the ideas behind it emerged in the beginning of the 20th century, the field only gained traction more recently with the introduction of modern sequencing techniques, which enabled the analysis of epigenetic factors on a genome wide scale.
Epigenetics is a multilayered net of regulation. Cytosine modifications of DNA at gene promoters leads to repression of gene expression. This DNA methylation, in turn, leads to the recruitment of chromatin modifying enzymes which modify histone tails. Those histone tails are modified most frequently at lysine residues which can be read, written, and erased by specialized enzyme complexes and this modification leads to activation or deactivation of a given chromatin region.
In another layer of epigenetic regulation, internal 6-methyladenine (m6A) residues in mRNA molecules influence the half-life of the mRNA and hence the levels of a given protein. Non-coding RNAs (miRNA, siRNA, piRNA, snoRNA, lncRNA) act as regulators of gene expression. Both miRNA and siRNA function in post-transcriptional RNA silencing pathways by base pairing with target mRNA, leading to degradation of mRNA and gene silencing. On the other hand, lncRNAs function in the process of X-inactivation and ensure the silencing of one of the two X chromosomes in female mammals.
All of these epigenetic factors are crucial for regulating normal organism development and homeostasis and provide a means for sensing and reacting to environmental changes. These epigenetic modifications can be passed on to future generations.
Numerous functional studies as well as genome-wide mapping of epigenetic marks and chromatin modifiers have revealed the importance of epigenomic mechanisms in human pathologies, including cardiovascular disease, developmental disabilities, and cancer.
Wherever the balance between epigenetic factors is disturbed, catastrophic consequences can result. New studies are looking for ways to identify aberrant epigenetic factors in liquid biopsies with the hope of early diagnosis and possible therapeutic intervention.
About the author

Stefan Dillinger, Ph.D.
Stefan was born in the Free State of Bavaria, Germany. After studying biochemistry in Ulm and Regensburg, he got his Ph.D. in the field of epigenetics, studying the distribution of heterochromatin around nucleoli during cellular senescence. As a graduate student he started his own German science podcast “The Random Scientist” and is now the host of Active Motif’s Epigenetics Podcast. When Stefan is not working at Active Motif or recording podcasts, he is a passionate runner (he finished the New York City Marathon in 3 hours 21 minutes!!) and loves to spend time with his wife and son.
Contact Stefan on LinkedIn with any questions, or to get running advice.
