<< Back to MOTIFvations Blog Home Page
Top Epigenetics Breakthroughs of 2018

January 25, 2019
2018 has come and gone, but before we focus too much on making plans and resolutions for 2019, let’s take a few minutes to look back at all the great epigenetics and gene regulation research that was published in 2018.
Epigenetics Research is at an All-Time High
Epigenetics research really started taking off in the early 2000’s, and according to PubMed, last year marked the largest number of papers published focusing on this fascinating research area.
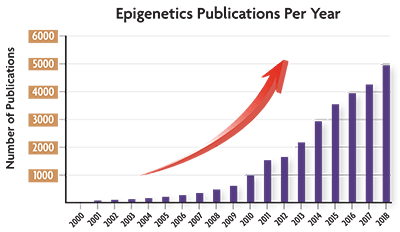
(Source data: PubMed)
There were so many great epigenetics papers last year that it was hard to keep up, so we wanted to highlight some of the most impactful and interesting research articles that we read in 2018. We aren’t able to cover all the great research published last year, but we’ll discuss a wide range of topics, including the most interesting applications of epigenetics research, novel epigenetic mechanisms of gene regulation, and new approaches and methods to investigate epigenetics in your research. Make sure to contact us to let us know what great scientific publications we missed, and we’ll try to highlight them in the future!
New & Improved Techniques for Epigenetics Research

Jump To: Novel Epigenetic Mechanisms | Interesting Epigenetic Applications
As the field of epigenetics research advances, better tools and techniques are needed to allow scientists to make their next great discovery. Many researchers, including the scientists at Active Motif, are heavily invested in developing new ways to study epigenetics. Some of the most important areas of epigenetic innovation are new approaches for single-cell analyses and higher throughput genomic and epigenomic assays with increasingly higher resolution.
Perturb-ATAC: Single-Cell CRISPR Screening + Epigenomic Profiling = Next Level Data
In a recent issue of Cell, scientists from the Stanford University School of Medicine present a novel method for epigenomic profiling at single-cell resolution, which they term Perturb-ATAC. This new high-throughput genetic screen was applied to determine the roles of diverse sets of trans-regulatory factors, including transcription factors, chromatin modifiers, and ncRNAs in the regulatory landscapes of different cell types.
The Perturb-ATAC approach combines multiplexed CRISPR interference or CRISPR knockouts of different key regulatory factors with analysis of chromatin accessibility variations within single cells. Individual cells are captured in chambers on their microfluidic system and then are subjected to lysis and Tn5 transposition. After transposition, CRISPR sgRNAs or sgRNA-identifying barcodes are used to create single-cell sequencing libraries.
According to the authors of this report, “Perturb-ATAC is a powerful and general strategy to dissect gene regulatory networks in development and disease.”
While this paper was technically officially published in 2019, it was e-published ahead of print in December 2018, so we snuck in on to our list of 2018 breakthroughs.
Reference: Rubin, A.J. et al. Coupled Single-Cell CRISPR Screening and Epigenomic Profiling Reveals Causal Gene Regulatory Networks. Cell 176, 361-376 (2019).
Link
Methylscape: Exploiting the Biophysical Properties of DNA Methylation to Serve as a Universal Cancer Biomarker
The epigenome in cancer cells is generally very distinct from non-cancer cells. The DNA methylation profile in cancer cells contain overall lower levels of DNA methylation (hypomethylation) relative to normal non-cancer cells, but there are also specific regulatory regions in the cancer cells that are hypermethylated in the cancer cells. These stark differences in DNA methylation patterns allow the potential to epigenetically identify cancer cells through simple, quick, and inexpensive methods.
New research led by scientists at the University of Queensland in Australia has developed a new method to probe the “Methylscape” of genomic DNA and quickly identify whether DNA methylation patterns seen in cancer cells are present. Their method stands out from other similar approaches to screen for DNA methylation-based biomarkers because it relies on the differential biophysical properties of methylated DNA between cancer and non-cancerous cells, rather than DNA sequencing or other approaches that directly detect the presence or absence of methyl groups on DNA.
The researchers found that the differences in the Methylscapes in cancer and normal cells have differential adsorption profiles to gold surfaces and nanoparticles which they exploited to develop assays to detect binding through either electrochemical or colorimetric techniques. The resulting assays are rapid (<10 minutes), sensitive (require low DNA input), and are easy to run (no special sample prep or processing steps), making this technology an attractive potential method as a first line test for cancer in non-invasively collected samples like blood, especially suited for use in the field outside of a hospital or laboratory environment, and in developing countries.
Reference: Sina, A.A., et al. Epigenetically reprogrammed methylation landscape drives the DNA self-assembly and serves as a universal cancer biomarker. Nat Commun. 9, 4915 (2018).
Link
EpiTOF: How Aging Influences Epigenetics of Immune Cells
They say that age is just a number. New research at Stanford University School of Medicine has now linked changes in levels of epigenetic modifications with aging. This report in Cell discussed the development of EpiTOF, a multiplexed mass cytometry assay that can investigate the global levels of 40 different histone modifications in T cell samples at single-cell resolution.
The researchers found that increased age was associated with increased heterogeneity in levels of histone modifications in T cells both between individuals as well as increased cell-to-cell variability within individuals. Similar results were seen in identical twins as well, which suggests that these variations in epigenetic profiles were likely due to environmental factors accumulating over time rather than genetic factors.
Reference: Cheung, P. et al. Single-Cell Chromatin Modification Profiling Reveals Increased Epigenetic Variations with Aging. Cell 173, 1385-1397 (2018).
Link
New Epigenetic Methods Reviewed: More & Better Epigenomic Data from Fewer Cells
Gone are the days where you could publish a paper in a high-profile journal with data on a single epigenetic modification at a single gene. Many current reports in the top tier journals investigate multiple epigenetic marks on a genome-wide scale, and increasingly at the single-cell and even single-molecule resolution.
This publication in Nature Genetics highlights many of the recent technical innovations in epigenetic analyses. Many of these new methods tend to be complicated and are still only accessible to a few labs worldwide, but we’re excited for the future and Active Motif will be leading the charge when it comes to making the latest techniques for studying epigenetics and gene regulation available to all researchers.
Reference: Shema, E., Bernstein, B.E., and Buenrostro, J.D. Single-cell and single-molecule epigenomics to uncover genome regulation at unprecedented resolution. Nature Genetics 51, 19-25 (2018).
Link
Epigenetics & ChIL – New Method Offers an Alternative to ChIP-Seq
ChIP-Seq assays have revolutionized the way researchers could interrogate epigenetic modifications and transcription factor binding on a genome-wide scale. ChIP-Seq does have some limitations though. Most notably, it is often difficult to immunoprecipitate enough material from low cell numbers to generate high-quality next-generation sequencing data.
A group of researchers in Japan rose to the challenge and developed a new immunoprecipitation-free method to investigate genome-wide epigenetic profiles from low numbers of cells. Their technique is called “ChIL”, which stands for Chromatin Integration Labelling. Rather than performing a chromatin preparation followed by an IP reaction, ChIL instead uses a primary antibody for the target of interest followed by addition of the ChIL probe. A ChIL probe is a secondary antibody that recognizes the first antibody conjugated to DNA sequences required for amplification and library preparation and the Tn5 transposase. The ChIL probe essentially carries out a tagmentation-like library prep reaction at the sites bound by the primary antibody which can be amplified, purified, and sequenced.
The ChIL-Seq method was validated to work well with low cell numbers, even down to single cells, for several epigenetic modifications such as H3K4me3 and H3K27Ac. Although the researchers that developed ChIL acknowledge that this method does have some drawbacks, mainly that it takes several days and requires many hands-on steps throughout the protocol, it’s exciting to see innovations to epigenomics technologies and we can’t wait to see what improvements will be coming next for ChIL-Seq.
Reference: Harada, A. et al. A chromatin integration labelling method enables epigenomic profiling with lower input. Nat Cell Biol. Epub ahead of print. (2018)
Link
Novel Epigenetic Regulatory Mechanisms

Jump To: New & Improved Epigenetic Techniques | Interesting Epigenetic Applications
While developing new technologies for investigating epigenetic modifications is important, innovation can’t stop there. The next category of epigenetic breakthroughs we’re highlighting are those that discovered new mechanisms of epigenetic regulation of gene expression.
RNA Methylation is a New Player in Epigenetic Regulation of the Stress Response
Although there are over 100 epigenetic modifications to RNA, N6-methyladenosine (m6A) is the most abundant and has therefore been in the spotlight the last few years. The biological roles of m6A are still being clarified, but it appears to be enriched in untranslated regions (UTRs) of the transcriptome and its levels and localization change during several cellular processes such as stem cell differentiation, tumorigenesis, and the stress response.
In a recent issue of the journal Neuron, Mareen Engel and colleagues published the results of their study examining the levels of m6A and N6,2′-O-dimethyladenosine (m6Am), which together the authors referred to as m6A/m, during the stress response in adult mouse brain tissue. The researchers performed genome-wide m6A/m-RIP-Seq assays and found both brain region-specific and gene-specific changes in m6A/m. These findings indicate that epigenetic modification of RNA represents a novel layer of complexity in gene expression regulation after stress and that dysregulation of the m6A/m response may contribute to the pathophysiology of stress-related psychiatric disorders.
Reference: Engel, M. et al. The Role of m6A/m-RNA Methylation in Stress Response Regulation. Neuron 99, 389–403 (2018).
Link
Cytidine Acetylation in mRNA - New RNA Epigenetics Flavor of the Month?
Continuing with the RNA modification theme, another epigenetic modification of RNA, N4-acetylcytidine (ac4C), has been identified as a new modification to mRNA catalyzed by the enzyme N acetyltransferase 10 (NAT10). The ac4C modification is mainly found on transfer RNA and ribosomal RNA molecules, but recent reports have hinted at this modification also being present in mRNAs.
In a recent Cell paper, Daniel Arango and colleagues performed several assays to investigate the presence and role of ac4C in mRNA, including ac4C-RIP-Seq in both WT cells and in cells with NAT10 knocked out with CRISPR. They found >4,000 peaks in mRNAs containing the ac4C modification, and it was preferentially present in coding sequences and 5’ UTRs, and less abundant in 3’ UTRs. Further studies with the NAT10 KO cells suggested that ac4C plays a role in promoting mRNA stability and translation efficiency.
The paper represents the first report of ac4C in mRNA molecules, and highlights both the importance of RNA epigenetics and how little of this area we currently understand about RNA modifications. The authors wrap up their paper by speculating that there might be an “epitranslation” code with RNA modifications that is analogous to the post-translational modifications of histones that have become the widely accepted “histone code” model of epigenetic regulation of gene expression.
Reference: Arango, D. et al. Acetylation of Cytidine in mRNA Promotes Translation Efficiency. Cell 175, 1872-1886. (2018).
Link
Histidine Methylation: Uncovering a New Epigenetic Role for SETD3
Two studies published in December 2018 revealed that the H3K4/H3K36 methyltransferase SETD3 is responsible for methylation of β-actin H73. Methylation of histidine has previously been observed in several proteins, but its function remains enigmatic. These studies represent the first evidence of a histidine methyltransferase identified to date in any species.
Through the use of SETD3 knockout mice and HAP1 knockout cells the authors of these two studies convincingly demonstrated that SETD3 is the sole enzyme responsible for methylation of β-actin histidine 73. Quantitative proteomics performed by Dr. Wilkinson and colleagues additionally reveal that β-actin H73 is the main physiological substrate of SETD3 in mouse cells.
Dr. Kwiatkowski and colleagues additionally demonstrated that SETD3 depletion resulted in reduced actin filament stability and compromised cytoskeleton integrity. The increased depolymerization observed in cells with hypomethylated F-actin also appears to drive a metabolic shift towards glycolysis. Furthermore, Dr. Wilkinson’s group also found that loss of SETD3 has profound effects on smooth muscle contraction, and that the enzyme is particularly important for uterine contractions during childbirth.
References: Wilkinson, A. et al. SETD3 is an actin histidine methyltransferase that prevents primary dystocia. Nature 565, 372-376 (2018).
Link
Kwiatkowski, S. et al. SETD3 protein is the actin-specific histidine N-methyltransferase. eLife 7:e37921 (2018).
Link
Role for Chromatin Remodeling Complexes in Cancer
While post-translational modifications of histones could probably be considered the “rock stars” of epigenetics, it’s really how those modifications change the biochemical and structural properties of chromatin that’s where the magic happens. For example, euchromatin modifications like H3K4me3 and H3K27Ac that are associated with active transcription tend to open the chromatin to make it more accessible to transcription factors and also play a role in the direct recruitment of transcription activators and other proteins involved in turning genes on. On the other hand, heterochromatin modifications like H3K27me3 facilitate the chromatin packing up tightly to repress gene expression by preventing access to transcription factors.
A class of enzymes referred to as chromatin remodelers adds another layer of regulation by physically moving (or removing) nucleosomes within the genome to either promote or repress gene expression and other biological processes.
A recent paper by Brittany Michel at the Dana Farber Cancer Institute and Harvard Medical School and her colleagues and collaborators investigated the genome-wide localization of a non-canonical SWI/SNF chromatin remodeling complex called ncBAF and found that it primarily associated with genomic loci bound by the chromatin insulator protein CTCF and promoter regions. They took their study further and showed that ncBAF is a synthetic lethal target in certain cancers, suggesting that it may be a therapeutic candidate to target for treatment of cancers such as synovial sarcoma and malignant rhabdoid tumors driven by SWI/SNF mutations.
Reference: Michel, B.C. et al. A non-canonical SWI/SNF complex is a synthetic lethal target in cancers driven by BAF complex perturbation. Nat. Cell. Biol. 20, 1410-1420 (2018).
Link
Three-Dimensional Organization of Promoters and Enhancers
Genes in chromosomes of metazoan organisms are generally organized together into distinct domains or “units” that are essentially separate from surrounding chromatin contexts and these insulated domains can interact with other regions of the genome through long-range looping. Investigating these long-range interactions between different chromosomal loci is a hot area of research and increasing access to technologies such as Hi-C is allowing more researchers to start learning more about these interactions and their roles in biology.
A new paper in Molecular Cell from researchers at UCLA tackled the issue of 3-dimensional chromosomal organization of a type of chromosomal domain termed “insulated neighborhoods” using data from existing mouse genomic studies, such as Hi-C, 4C, and ChIP-Seq. They identified the insulated neighborhoods using bioinformatic approaches and also disrupted enhancer activity by knocking down a required enhancer factor called Esrrb using RNAi.
The main finding from this project was that enhancers generally co-regulate groups of genes in the insulated domains, without significantly affecting nearby gene outside of the insulated domain, suggesting that these higher order groups of genes are likely functional regulatory regions with distinct biological functions.
Reference: Sun, F. et al. Promoter-Enhancer Communication Occurs Primarily within Insulated Neighborhoods. Molecular Cell 73, 250-263. (2018).
Link
Cool and Interesting Epigenetic Applications

Jump To: New & Improved Epigenetic Techniques | Novel Epigenetic Mechanisms
While developing new technologies to learn more about the epigenetic landscape in biological samples and discovering novel epigenetic mechanisms are important, it’s how these technologies are used to answer biological questions that’s even more interesting.
Nature vs. Nurture: Dutch Winter Research Project Sheds Light on Epigenetics of Diet & Hunger
Increased understanding of epigenetic mechanisms of gene regulation has changed the nature vs. nurture conversation to become "what are the relative influences of nature (genetics) and nurture (environmental factors and epigenetics)?" It is well established that certain environmental changes and behaviors can result in changes to an organism’s epigenome, and sometimes these epigenetic changes are heritable, but the precise details and underlying mechanisms are not fully understood.
A landmark study published in early 2018 by a group led by researchers in the Netherlands attempted to address the relationship between DNA methylation and body mass index (BMI), serum triglycerides (TG), and glucose concentrations by performing genome-wide DNA methylation analysis in whole blood samples on a cohort of >400 individuals born to women that experienced extreme famine during the Dutch famine of 1944–1945 and another similar group born to siblings that did not experience the famine.
The Dutch famine occurred towards the end of World War II, when the German forces created a blockade that prevented food, fuel, and other resources from reaching small towns and farming villages in occupied regions. An estimated ~20,000 people died from famine during this period and another 4.5 million were affected but survived.
That travesty of humanity resulted in perhaps a small silver lining because of the follow-up studies performed on the impacted families. A large cohort study was done on the females affected by famine and children they gave birth to in the year or so following the end of the famine.
Recent epigenetic analysis of the Dutch famine cohort provided significant data supporting the hypothesis that epigenetic changes are influenced by non-genetic factors such as extreme malnutrition, and that short-term epigenetic changes can influence long-term health.
In particular, the researchers found that DNA methylation mediated BMI and TG changes, but not glucose concentrations. Furthermore, several genomic regions containing genes that encoded factors involved in metabolism and lipid regulation contained differential levels of DNA methylation, drawing a link between prenatal environmental conditions and the long-term risks of developing metabolic disorders in adulthood.
Reference: Tobi, E.W. et al. DNA methylation as a mediator of the association between prenatal adversity and risk factors for metabolic disease in adulthood. Sci Adv. 4, eaao4364 (2018).
Link
How Similar Are Identical Twins, Epigenetically Speaking
In a recent issue of Genome Biology, researchers examined monozygotic twins to try to identify regions of "epigenetic supersimilarity", which is defined as genomic regions at which the epigenetic similarity of monozygotic twins is substantially greater than can be explained by their genetic identity.
Using publicly available data of DNA methylation on CpG islands, the researchers located “super-similar” epigenetic loci in monozygotic twins, mostly located in sub-telomeric regions. Methylation at these loci was found to be stable from embryonic stages through adulthood and was associated with risk of developing several types of cancer. This study revealed a new level of epigenetic similarity in monozygotic twins, which is connected to human disease epidemiology.
Reference: Van Baak, T.E. et al. Epigenetic supersimilarity of monozygotic twin pairs. Genome Biology. 19, 2 (2018).
Link
Using Epigenetics to Learn How Songbirds Memorize New Tunes
Studying how songbirds like zebra finches learn new songs is a great way to better understand how organisms learn complex behaviors during development. It is well accepted that certain types of behaviors can only be learned during relatively short phases of development referred to as critical periods (CPs). Since the genomic sequences don’t change during development, epigenetics can likely offer an explanation for phenomena related to developmental learning.
In zebra finches, the juvenile males learn songs from adult males, which are referred to as “tutors” (interestingly, female zebra finches are not able to sing). Although zebra finches hear the songs from the adult males every day, they are only able to memorize songs during a short window that spans 30-65 days post-hatching. However, the environmental stimuli that the hatchlings experience influence their learning potential. If the birds are not exposed to a tutor male, their window to continue learning songs is extended. This developmental plasticity was assumed to be due to epigenetic changes, and this recent report confirmed that hypothesis.
In collaboration with the London lab from the University of Chicago, researchers from Active Motif used ChIP-Seq, RNA-Seq, and other molecular biology techniques to identify epigenetic changes in the forebrain of birds that were either exposed to a male tutor as in nature during development or that were isolated from tutors. Their experiments identified differences in epigenetic profiles and patterns of gene expression in the different bird populations, suggesting that exposure to a tutor results in epigenetic changes that contribute to the ability to learn and memorize the song behaviors. More broadly, this study provides additional evidence to support the idea that epigenetic mechanisms play important roles in critical period-based learning and development.
Reference: Kelly, T.K. et al. Epigenetic regulation of transcriptional plasticity associated with developmental song learning Proc. Biol. Sci. 285 (2018)
Link
Epigenetics to the Rescue for Cell-Based CAR T Therapy
Cell-based therapies such as CAR T represent one of the most important medical advancements in recent history. Being able to engineer cells to fight off cancers or other disease cells has the potential to redefine medicine and finally usher in the promise of truly personalized medicine that we’ve been waiting for since the idea started emerging decades ago.
As promising as CAR T therapy is, there are still significant technical hurdles that must be overcome. A recent Nature publication describes a case in which CAR T cell therapy was used to treat chronic lymphocytic leukemia (CLL). The researchers found that the majority of engineered cells that successfully treated CLL were derived from cells in which insertion of the CAR transgene interrupted the TET2 gene. TET2 is involved in the regulation of DNA methylation levels, suggesting that an epigenetic mechanism was responsible for this observation.
The researchers followed up their initial discovery by intentionally disrupting the TET2 gene and they were able to reproduce their previous observation of increased treatment potency. These results emphasize the importance of considering epigenetic targeting and regulatory mechanisms to improve the efficacy of cell-based therapies.
Reference: Fraietta, J.A. et al. Disruption of TET2promotes the therapeutic efficacy of CD19-targeted T cells. Nature 558, 307-312 (2018).
Link
2018 was a Great Year for Epigenetics Research, But the Best is Yet to Come!
Epigenetics research made great strides in 2018. There were many new assays developed to investigate epigenetics in different ways and using less sample, many new breakthroughs regarding the basics of how epigenetic mechanisms regulate gene expression and other cellular processes, and tons of great biological discoveries made possible by epigenetics research.
As we’re settling into 2019, it’s fun to think about what will come next. What breakthroughs will be made in the field of epigenetics this year?
A good guess would be that more new technologies will be developed that enable genome-wide assays like ChIP-Seq to be performed on a high-throughput scale. The sensitivity of epigenetic assays will also continue to improve, allowing more research to be performed on single cells and on precious clinical samples. Researchers will also almost certainly discover new epigenetic modifications and new roles for previously identified modifications.
We’ll be keeping our eyes and ears open for all the latest breakthroughs, so stay tuned!
<< Back to MOTIFvations Blog Home Page



