<< Back to MOTIFvations Blog Home Page
Complete Guide to Understanding and Using ATAC-Seq

February 9, 2021
Methods like chromatin immunoprecipitation and reduced representation bisulfite sequencing have made it easier for researchers to investigate epigenetic modifications on a genome-wide scale. However, one of the potential limitations of these methods is that you need to already have an idea about what epigenetic mechanisms are at play in your experimental system.
The ATAC-Seq method addresses this issue by giving researchers information about chromatin accessibility across the genome, regardless of the mechanism responsible. Many researchers have started using ATAC-Seq as a first-pass screening approach to identify changes in chromatin accessibility between samples, and then they can focus their follow-up experiments based on the results.
This article discusses what ATAC-Seq is, it’s history, how it works, and some discoveries enabled by ATAC-Seq.
What is ATAC-Seq?
ATAC-Seq stands for Assay for Transposase-Accessible Chromatin with high-throughput sequencing. The ATAC-Seq method relies on next-generation sequencing (NGS) library construction using the hyperactive transposase Tn5. NGS adapters are loaded onto the transposase, which allows simultaneous fragmentation of chromatin and integration of those adapters into open chromatin regions. The library that is generated can be sequenced by NGS and the regions of the genome with open or accessible chromatin are analyzed using bioinformatics.
The main advantages of ATAC-Seq compared to other techniques, such as FAIRE-Seq or DNase-Seq that investigate similar chromatin features, are the lower number of cells that are required for the assay and the relative simplicity of its two-step protocol.
History of ATAC-Seq
The ATAC-Seq method was first published in 2013 in the journal Nature Methods by lead researcher Jason Buenrostro in the labs of Howard Chang and William Greenleaf at Stanford University.
At that time, they were looking for an alternative to current methods that were used to study open chromatin, nucleosome positioning, and transcription factor occupancy. The previous methods that were available all had several limitations including the need for a high amount of starting material, complex and time-consuming protocols, and the lack of ability to simultaneously assess the three chromatin mechanisms mentioned earlier together in one assay.
The team of researchers set out to overcome the limitations of current methods and came up with an assay that was able to analyze the overall epigenetic profile from a significantly lower number of cells than the other techniques.
Their method relied on the hyperactive Tn5 transposase that was already being used for tagmentation-based NGS library preparation methods, such as the Nextera approach developed by Epicentre (which was later acquired by Illumina). The tagmentation process can be used to fragment genomic DNA in vitro and simultaneously add adapters for high-throughput sequencing. The authors hypothesized that if a similar approach was used in vivo, the addition of the adapters would mainly take place in open chromatin regions, where no steric hindrance of the transposase would occur, allowing the enzyme to preferentially access these regions.
In their proof of concept study, they investigated unfixed nuclei isolated from populations of 500 and 50,000 cells. The data they generated were very similar to DNase-Seq and FAIRE-Seq, both of which require 1-50 million cells, demonstrating that the ATAC-Seq method represented a significant improvement over those older methods in terms of the amount of sample required.
Furthermore, since the transposase only adds the NGS adapters to accessible DNA, ATAC-Seq offers an additional benefit: it can also be used to generate a high-resolution map of nucleosome positions as well as transcription factor binding profiles. The relatively short two-step protocol suggested to the researchers that ATAC-Seq could also potentially be used to generate personal epigenetic profiles from clinical samples.
Hear the story of how Jason Buenrostro from Harvard University tried something new in the lab and in the end developed the ATAC-Seq method, on our Epigenetics Podcast
How Does ATAC-Seq Work?
As mentioned previously, one of the main advantages of ATAC-Seq over other methods which analyze accessible chromatin regions or nucleosome positioning, like DNase-Seq or FAIRE-Seq, is that ATAC-Seq can be performed with significantly fewer cells (~ 50,000 cells for ATAC-Seq compared to millions of cells for the other methods). The historical methods are also more laborious compared to ATAC-Seq, which can be completed in a two-step process within several hours.
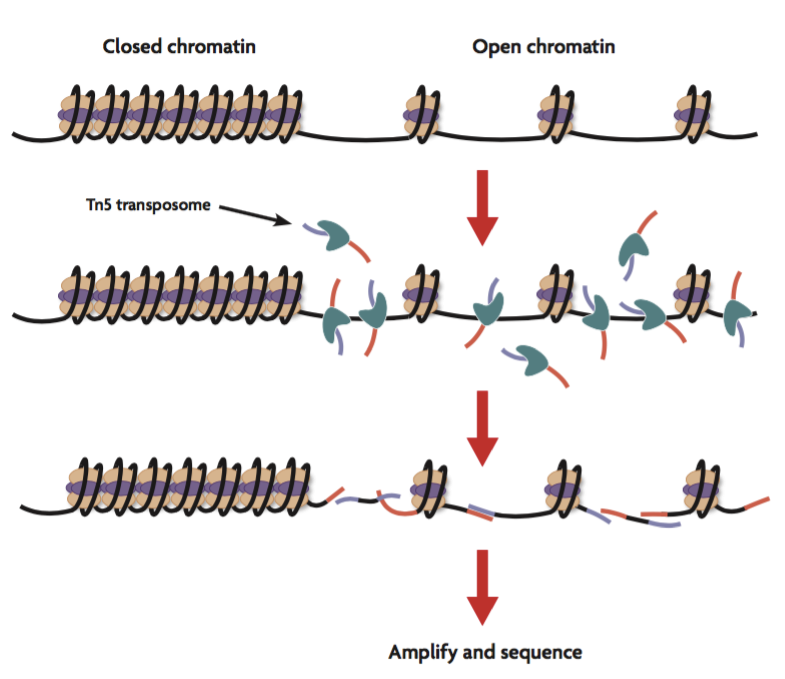
Below, we give a short overview of the ATAC-Seq protocol, which can be divided up into two main parts: cell preparation and transposition.
Cell Preparation
As a first step, the cells need to be harvested. Since the number of cells used in ATAC-Seq assays is crucial for the transposition reaction and size distribution of the generated DNA fragments, counting the cells is important. Furthermore, cells should be intact and in a homogeneous, single-cell suspension. After harvesting, cells are lysed with a nonionic detergent to yield pure nuclei.
Transposition Reaction
The resulting chromatin is then fragmented and simultaneously tagmented with sequencing adapters using the Tn5 transposase to generate the ATAC-Seq library. After purification, the library can be amplified by PCR using barcoded primers. The resulting library can then be analyzed by qPCR or next-generation sequencing.
Additional ATAC-Seq Considerations
It is important to use the right number of Cells for the transposition step. The general recommendation is 25,000-75,000 cells. Using too few or too many cells can lead to over- or under-digestion, which would lower the quality of the library. Furthermore, fixation and harsh mechanical shearing of the cells tend to reduce the quality of the data.
Recent ATAC-Seq Applications & Breakthroughs
Through its short two-step process, ATAC-Seq has proven applicable for various cell types and model systems. The number of papers that were published using ATAC-Seq since the method was published in 2013 has doubled every year. We describe below some publications from different fields that show the impact and applicability of this method.
ATAC-Seq & Cancer
A team of researchers in Miami and their collaborators used ATAC-Seq in the growing field of cancer epigenetics to investigate the effect of the Polycomb repressive complex 1 (PRC1) on chromatin states in breast cancer. ATAC-Seq experiments showed that depletion of RING1B, a member of the PRC1 complex, leads to a change in chromatin accessibility at enhancer regions. Furthermore, the loss of RING1B led to the loss of about 500 accessible chromatin region peaks and the appearance of more than 600 new open regions near enhancers. Motif analysis showed that peaks lost at enhancer regions contained FOXA1/2-binding sites, whereas de novo peaks contained CTCF-binding sites, indicating a dual role for RING1B in breast cancer cells.

Investigating Aging with ATAC-Seq
Age-related macular degeneration (AMD) is a medical condition that leads to irreversible reduced vision. While it is known that several genetic loci contribute to increased risk for this disease, the role of epigenetic factors is largely unknown.
Scientists at Johns Hopkins University School of Medicine used ATAC-Seq to study genome-wide chromatin accessibility in AMD versus control patients. Their results demonstrated that chromatin accessibility was heavily decreased in the retina and retinal pigment epithelium (RPE) from AMD patients. Further analysis of the data showed that specific transcription factors and cellular functions were enriched in differentially accessible chromatin regions.
Moreover, they studied potential environmental effects such as cigarette smoke on AMD. Treating terminally differentiated iPSC-derived RPE cells with cigarette smoke induced a decrease in chromatin accessibility that resembled the pattern that was observed in AMD patients.
The researchers concluded their study by trying to identify potential drug targets for the treatment of AMD. Analysis of RNA-Seq data revealed that HDAC11, a histone deacetylase, was overexpressed in AMD and could be a possible target for future therapies.
ATAC-Seq & Immunology
During the maturation of B cells in the humoral immune response, B cell progenitors need to undergo massive phenotypic changes that are mediated by a multilayered chromatin organization that involves the coordination of multiple transcription factors. A recent study published in Nature Immunology by a group of researchers at Cornell and their collaborators investigated the role of lysine-specific demethylase 1 (LSD1) in B cell development.
The team used ATAC-Seq to study the open chromatin regions in sorted viable germinal center B cells from Lsd1 mutant mice. They observed 733 regions that gained chromatin accessibility upon Lsd1 knock-out and 314 regions lost accessibility. There was also increased transcriptional activation associated with those 733 “opened” regions. Further experiments, including ChIP-Seq, revealed that BCL6, a transcriptional repressor important for coordination of enhancer and promoter pausing, directly binds the LSD1 protein and recruits it to chromatin, and this interaction is required to drive the malignant transformation of germinal center B cells.
Using ATAC-Seq to Study Cell Differentiation & Development
Cellular differentiation and lineage commitment are often mediated by epigenetic mechanisms, including large changes in chromatin structure and gradual transition from a generally more open chromatin state in progenitor and stem cells to a more closed, compact chromatin state in differentiated cells.
In a paper published in the journal Science, a team of researchers in Israel investigated the chromatin states during blood cell development. They characterized the stages of differentiation in hematopoiesis by looking at four histone modifications (H3K4me1, H3K4me2, H3K4me3, and H3K27ac) at different stages of development of all the main blood cell lineages.
Using iChIP they were able to identify 48,415 enhancers and 90% of them changed state during hematopoiesis. While 60% of those enhancers showed the typical behavior in which they gradually get inactivated and only remain active in the relevant lineage, about 40% get activated de novo during differentiation.
These newly established enhancers contain the H3K4me1 modification, which is associated with an increase in chromatin accessibility. To measure whether the chromatin at these enhancers was more accessible, H3K4me1 and H3K27ac ChIP-Seq data were correlated with ATAC-Seq data. The researchers observed a closer correlation in the overlap of ATAC-Seq peaks with H3K4me1 compared to H3K27ac, indicating that the gain or loss of H3K4me1 at enhancers affects the formation of accessible chromatin. Furthermore, ATAC-Seq enabled the identification of new regulators of lineage specification in hematopoiesis.
In summary, ATAC-Seq allowed the scientists to clearly demonstrate that chromatin states are very dynamic during blood cell differentiation, and enhancers are not only deactivated as previously reported, but are also established de novo, which leads to a new model of massive dynamic reorganization of chromatin during differentiation and development.
Single-Cell ATAC-Seq (scATAC-Seq)
A major limitation of all methods that use populations of millions of cells is that the data they produce is always an average of what’s happening in each of the individual cells in the population, which averages out the cell-to-cell variability that might be present in the sample and therefore might eliminate the ability to make observations about interesting phenomena in sub-populations. This variability is often an important feature of biology when it comes to things like tumor heterogeneity or developmental processes. Jason Buenrostro and colleagues, the original inventors of the ATAC-Seq method, set out to improve ATAC-Seq by adapting the ATAC-Seq protocol to make it compatible with single-cell analyses, which they published in the journal Nature.
Their initial experiments used a microfluidics platform from Fluidigm to help them to investigate single cells, but many single-cell ATAC-Seq (scATAC-Seq) protocols today use the 10X Genomics platform. After the single cells were isolated, they are tagmented with the Tn5 transposase and the libraries are amplified via PCR with cell-identifying barcoded primers. After pooling those libraries, they can then be sequenced as usual.
In summary, this approach improves ATAC-Seq in a way that enables researchers to study the biological variation in many sample types at the single-cell level, which will lead to a greater understanding of the molecular foundation of cellular heterogeneity.

How to Get Started Using ATAC-Seq in Your Research
To get ATAC-Seq started in your lab you can consult us at Active Motif as we offer an end-to-end ATAC-Seq service. You send us your samples (cells or tissue) and get your analyzed data back in a matter of weeks. The ATAC-Seq service covers all steps in the protocol, including membrane permeabilization, the addition of the transposase carrying sequencing adaptors, transposase-mediated insertion of adaptors at accessible genomic locations, library amplification, next-generation sequencing on the Illumina platform, and bioinformatic analysis. We have made optimizations to the standard ATAC-Seq protocol to ensure that we will generate the highest quality data for you.
We accept cultured cell lines, primary cells, or frozen tissue samples, and we suggest submission of 100,000 cells or 20-50 mg of frozen tissue. The number of replicates is always a matter of debate, but when it comes to our ATAC-Seq service, we usually only recommend duplicates because we have shown that the reproducibility of the data we generate is excellent. However, ultimately you will decide on the best experimental design for your system.
The bioinformatics data analysis is performed by our in-house team of bioinformatics experts. Standard bioinformatics analysis includes:
- Sequence Analysis: Sequencing reads are mapped to the genome and duplicate reads are removed.
- Peak Finding: Both reads from the paired-end sequencing are used for peak calling using the peak caller MACS 2.1.0. No in silico NGS read extension is performed.
- Determination of Fragment Density: To identify the density of transposition events along the genome, the genome is divided into 32 bp bins and the number of fragments in each bin is determined. For this purpose, reads are extended to 200 bp to smooth the data.
- Normalization: The number of aligned reads of all samples is normalized by random sampling to contain the same number of reads present in the sample with the fewest aligned reads.
- Active Region Analysis: To compare peak metrics between 2 or more samples, overlapping peaks are grouped into “Active Regions”, which are a proprietary metric defined by the start coordinate of the most upstream peak and the end coordinate of the most downstream peak.
If you are working with complex or heterogeneous samples, our single-cell ATAC-Seq service is available and might be the best option to help you achieve your experimental goals.
Setting up ATAC-Seq assays in your lab is also an option. To get started performing ATAC-Seq yourself you first would need to get the preloaded Tn5 Transposase. After that, you would need to purify the nuclei from your cells or tissue and find the right conditions for the transposition reaction (e.g. the right ratio between chromatin and Tn5). After library preparation and amplification, next-generation sequencing needs to be performed and data need to be analyzed subsequently.
Active Motif also offers an optimized ATAC-Seq Kit that researchers can use to generate sequencing-ready ATAC-Seq libraries from cell or tissue samples. This kit contains the reagents needed to perform ATAC-Seq and a streamlined protocol to ensure best results.
While ATAC-Seq is much faster and easier than previous methods, it still presents challenges to many researchers that are not experienced with the protocol, leading scientists to increasingly turn to the epigenetics experts at Active Motif to generate high-quality data they can trust.

Summary: The ATAC-Seq Method Has Changed the Way Researchers Start Doing Epigenetics
When first getting started in the field of epigenetics, the vast variety of methods that are out there can be overwhelming. To pick one approach to tackle your biological question might not be so obvious or easy. Methods like DNase-Seq, FAIRE-Seq, DamID, Hi-C, or ChIP-Seq require millions of cells and often a fair amount of optimization, and in the end, they may only deliver a limited amount of information, leaving you with an incomplete picture of the epigenomic landscape in your samples.
ATAC-Seq has changed the way scientists can approach getting started with epigenetics. The ATAC-Seq protocol requires only about 50,000 cells as starting material, and with its relatively short two-step protocol, it is an attractive method to start your epigenetic journey.
Whether you want to analyze the state of the chromatin in your sample or compare the chromatin state before and after a special treatment, ATAC-Seq allows you to investigate genome-wide chromatin changes and can offer guidelines about which epigenetic modification or transcription factor should be studied next in the follow-up experiments and which method should be used to study them.
ATAC-Seq has made it easier and faster than ever to study the chromatin landscape and take your first steps into the world of epigenetics. New variations, including Omni ATAC-Seq (Omni-ATAC-Seq) and others are increasingly popular. Check back often to learn about new ATAC-Seq, CUT&Tag and other methods as they develop.
Contact us if you are interested in learning more about ATAC-Seq, how our service works, or to obtain a quote.
About the author

Stefan Dillinger, Ph.D.
Stefan was born in the Free State of Bavaria, Germany. After studying biochemistry in Ulm and Regensburg, he got his Ph.D. in the field of epigenetics, studying the distribution of heterochromatin around nucleoli during cellular senescence. As a graduate student he started his own German science podcast “The Random Scientist” and is now the host of Active Motif’s Epigenetics Podcast. When Stefan is not working at Active Motif or recording podcasts, he is a passionate runner (he finished the New York City Marathon in 3 hours 21 minutes!!) and loves to spend time with his wife and son.
Contact Stefan on LinkedIn with any questions, or to get running advice.
Related Articles
Guide to Generating the Best ChIP Data

March 15, 2019
The chromatin immunoprecipitation (ChIP) assay has become one of the most popular laboratory techniques to investigate the association of DNA-binding proteins and histones with chromatin. This article covers the major challenges of ChIP assays and how to overcome them to generate the best ChIP data.
Read More
Comprehensive Guide to Understanding and Using CUT&Tag Assays

March 9, 2020
CUT&Tag shows a lot of promise and has the potential to alleviate some ChIP limitations, but it also has its own set of limitations that must be considered. This article covers what CUT&Tag is and describes the advantages and drawbacks of this method.
Read More
<< Back to MOTIFvations Blog Home Page




