RRBS Service Overview
Reduced Representation Bisulfite Sequencing (RRBS) provides DNA methylation data at single base pair resolution at lower cost than Whole Genome Bisulfite Sequencing (WGBS) and with greater coverage and less bias than bead array-based DNA methylation platforms.
RRBS is a method designed to selectively isolate DNA containing CpGs by using DNA restriction endonuclease digestion and amplification of fragments smaller than 300 bp. In brief, methylation insensitive endonucleases are used to cut DNA fragments at CpG locations. Since CpG islands and promoters have a high density of CpGs they are cut at a high frequency and the fragments derived from these regions tend to be short. This subset of short fragments is preferentially amplified when the library is generated through PCR and additional selection occurs during sequencing cluster generation. The final fraction of DNA that is sequenced represents a small fraction of the total genome and is enriched in CpGs.
Active Motif is one of a few RRBS services commercially available that uses two restriction enzymes to generate DNA fragments with CpG ends. Two methylation insensitive endonucleases, Msp1 and Taq1, are used to cut C^CGG and T^CGG respectively. This strategy allows for integration of more CpG dense regions with more accuracy.
RRBS Highlights:
- DNA methylation data on up to 5 million CpGs per sample.
- Biologically relevant target regions including promoters and CpG islands.
- Single-end sequencing depth of >30,000,000 reads.
- Low input DNA requirement of 100 ng.
- Reduced library bias through removal of PCR duplicates by random bar code incorporation.
Deliverables:
- Raw data (FASTQ) output files
- Aligned data (BAM) files
- Methylation table includes percent methylation value for each CpG
- Differential methylation analysis
- Figures & graphs
– Includes coverage depth analysis, correlation plots, and pie charts

What our customers are saying about us
"We worked with Active Motif to process some FFPE and frozen liver samples sourced from 25+year old samples for an RRBS assay. The overall process of submitting samples, communicating with Active Motif, receiving the data, and obtaining the summary was quick and painless. The whole operation was smooth and professionally done."
Brian Chorley, PhD
Environmental Protection Agency
View complete list of testimonials >
RRBS Service Data
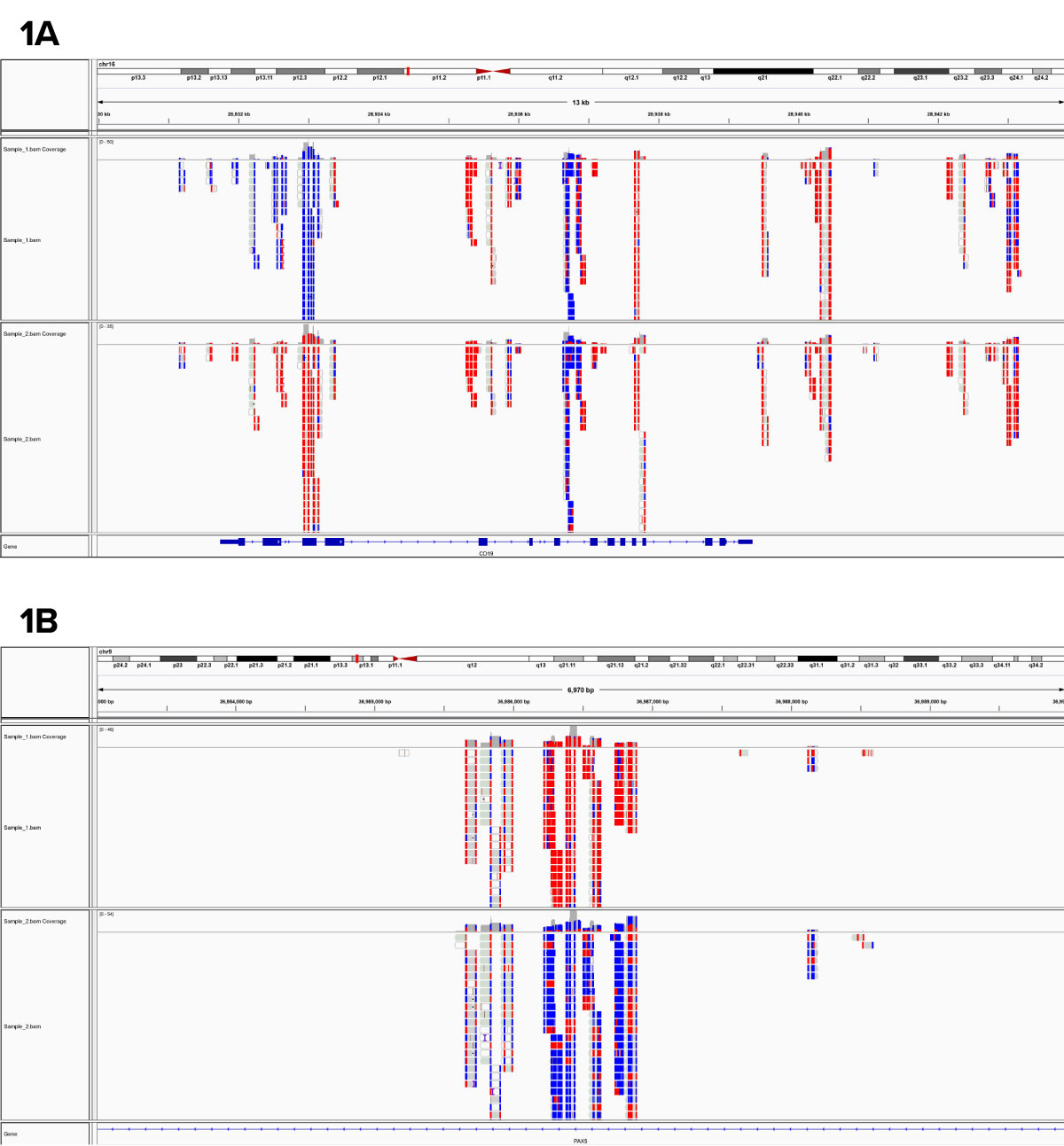
Figure 1: RRBS data from human samples.
The displayed regions are representative regions from the genome-wide data set and shows differential DNA methylation at 1A) an exon of CD19 and 1B) an intron of PAX5. Each block is a separate data point with red representing a methylated cytosine and blue representing an unmethylated base.
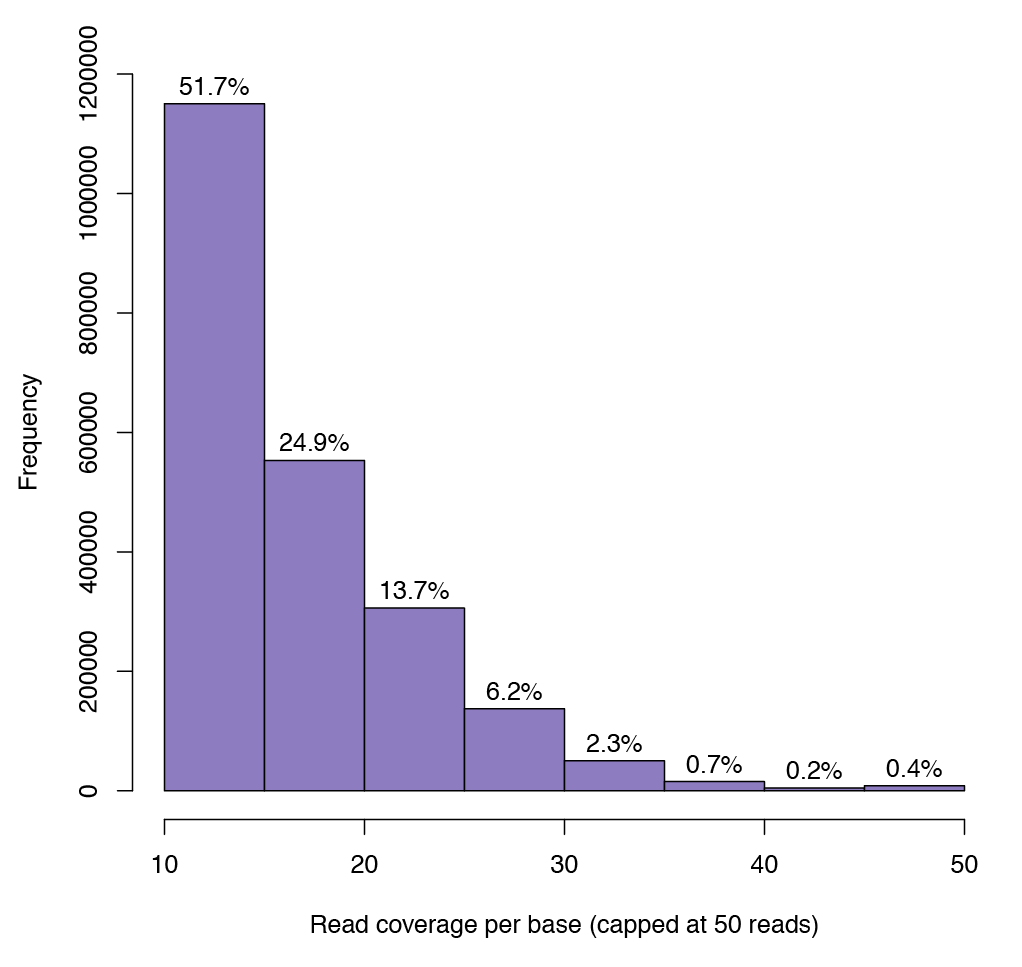
Figure 2: Graph illustrating sequencing depth coverage for any given CpG.
The minimum sequence depth for a CpG to be included in the analysis is 10. Over 1 million CpG sites have a coverage of at least 10 reads.
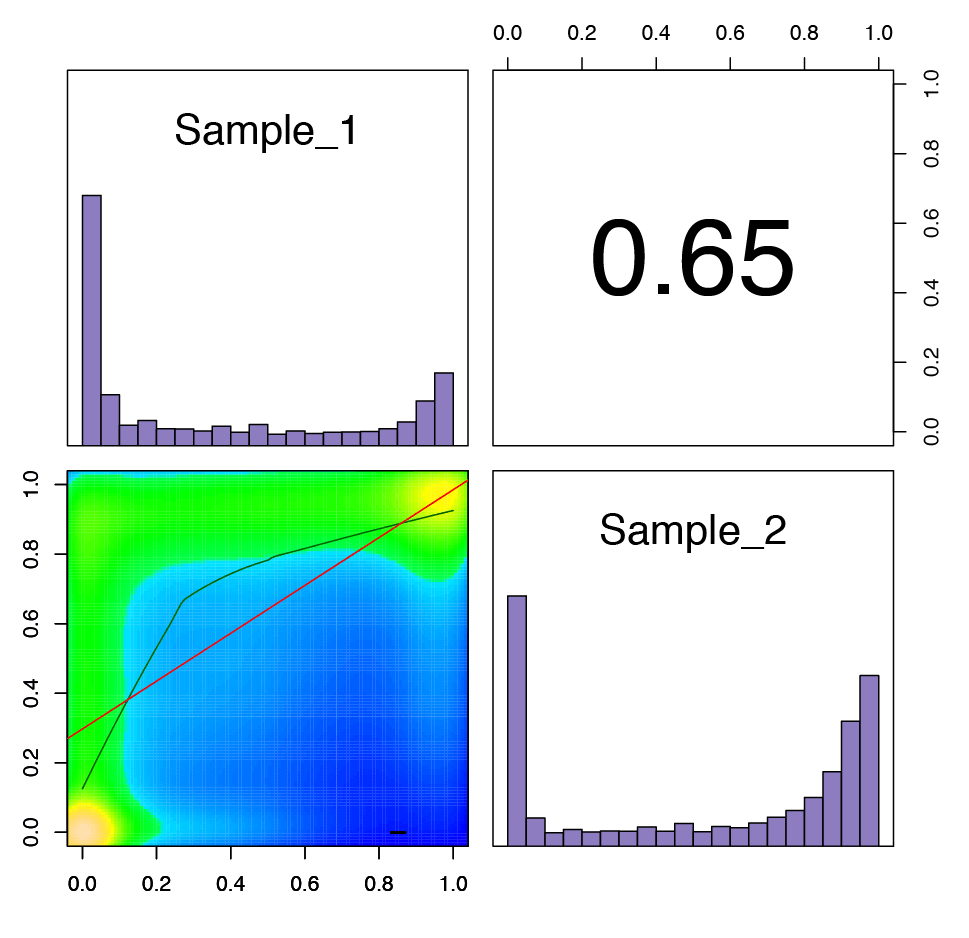
Figure 3: Graph illustrating the correlation between each sample analyzed.
The upper right corner shows the Pearson correlation coefficient for the two samples. The bar plots show the distribution of the percent methylation in each sample, with the majority of analyzed CpG sites having 0 or 100% methylation. The scatter plot in the lower left corner shows the level of methylation at shared CpG sites for each sample.
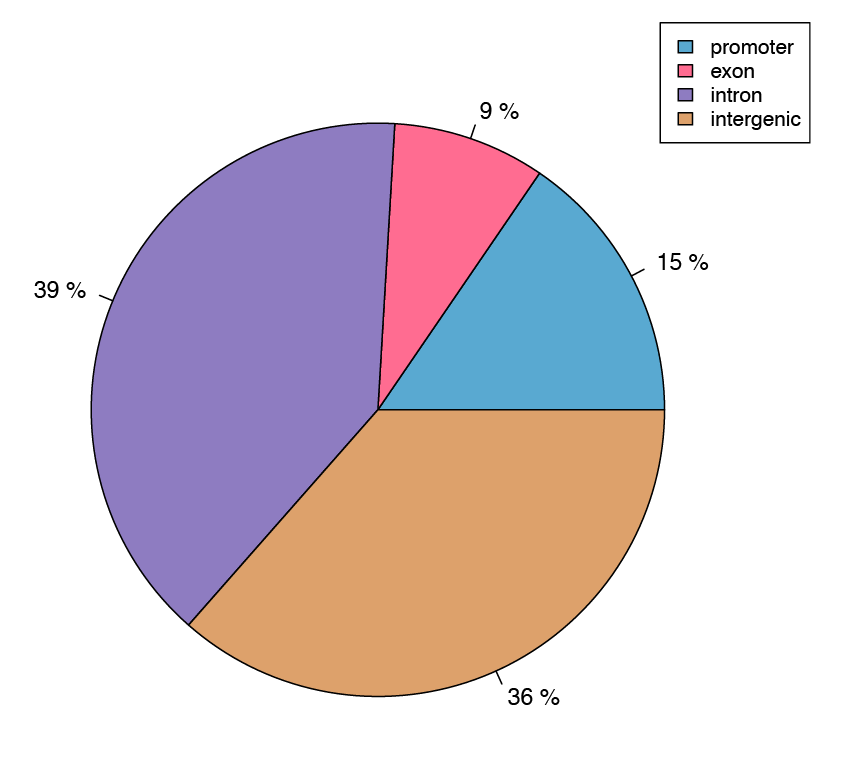
Figure 4: Pie chart showing the assignment of differentially methylated cytosines to genic features.
A differential methylation analysis was performed on a per CpG basis, and identified sites were then aligned to annotated gene features such as promoters and exons.
RRBS Service Publications
Search our database of customer publications that have used our RRBS services.
RRBS Service Documents
RRBS Service Sample Submission Portal
Our online sample submission portal allows you to easily upload your service project samples and track your project status. Follow the sample submission instructions in the portal to ensure that all your samples arrive at Active Motif in the best possible condition and properly associated with your project.