<< Back to MOTIFvations Blog Home Page
Using RIME to Analyze Protein-Protein Interactions on Chromatin

September 3, 2019
There are many different ways to identify the components of protein complexes to gain a better understanding of their roles in biological processes, each with their own set of benefits and limitations. The RIME method has gained popularity with scientists in recent years because of its low requirement for starting material relative to other methods and its sensitive and specific detection of protein-protein interactions without a need for any genetic manipulations.
This article covers what RIME is, the history of RIME, how RIME works, some recent discoveries made using RIME, and how to get started with RIME.
What is RIME?
RIME is an acronym for “Rapid Immunoprecipitation Mass spectrometry of Endogenous proteins”, and is a technique developed recently to identify transcriptional co-factors and chromatin-associated proteins. The core of the RIME method involves affinity purification of the protein of interest by bead-bound antibodies. The proteins that co-purify with the target protein of interest are then analyzed and identified by mass spectrometry following on-bead tryptic digestion.
The RIME method uses formaldehyde to crosslink proteins and DNA that are in very close proximity with each other, which makes it possible to identify and investigate transient and low-affinity protein-protein interactions. Formaldehyde is already the “go-to” fixative for methods like immunohistochemistry (IHC) and ChIP-Seq. Using formaldehyde in RIME protocols allows the same samples being used in ChIP-Seq assays, which identify protein-DNA interactions, be investigated by a proteomics approach, to study protein-protein interactions.
The benefits of RIME are:
- RIME works with small amounts of starting material relative to other immunoprecipitation followed by mass spectrometry (IP-MS) methods.
- The RIME assay is quick, the whole protocol can be completed within 2-3 days.
- RIME allows detection of low affinity and transient binding and minimizes detection of artificial interactions, providing the most relevant identification of protein-protein interactions.
- Transfections, epitope tagging, and other manipulations are not necessary because RIME works with endogenous proteins.
History of RIME and Recent Protocol Developments
The RIME method was first published in 2013 in the journal Cell Reports by lead researcher Hisham Mohammed and colleagues in the lab of Jason Carroll at Cancer Research UK and the University of Cambridge.
The team of scientists set out to find an alternative for methods that were used historically to capture protein complexes using antibodies coupled with mass spectrometry for the identification of protein-protein interactions. Those older approaches often involved using antibodies to immunoprecipitate proteins of interest that are overexpressed or epitope-tagged versions of the protein.
These early methods had some limitations because approaches that use overexpressed or tagged proteins suffer from influences like altered protein complexes or steric hindrance, making the results less biologically relevant.
On the other hand, previous methods that did not use overexpression systems or epitope tags often suffered from their inability to detect interactions that had a relatively low affinity or interactions that were dynamic or transient.
Therefore, Mohammed and colleagues wanted to develop a method that was sensitive and specific enough to identify both stable high-affinity protein-protein interactions and transient interactions in a complex of interest.
The First RIME Publications
The first publication of the RIME method described the purification of the estrogen receptor (ER), which is the main driving transcription factor in luminal breast cancer. The many advantages of RIME mentioned above made it possible to use limited, primary material, such as breast cancer samples to identify novel co-factors that associated with ER. The team of researchers used RIME to show that GREB1, a poorly-characterized protein, was recruited to chromatin by ER, where it activated transcription as an essential factor. GREB1 also stabilized the associations between ER and additional co-factors.
In the second RIME publication, which was published in 2015 in the journal Nature, a group of scientists in the Carroll lab focused on the interaction of the progesterone receptor (PR) and ER in breast cancer. The researchers were able to use RIME to show that activation of PR led to increased interaction between PR and the ER complex. This interaction was previously known, but not to the extent that was revealed in this publication.
The nature of this interaction is used as a clinical prognosis marker for breast cancer, with reduced PR expression indicating poor clinical outcome. The findings of this paper show that PR is not just a marker of an intact PR-ERα complex but is a critical component in this regulatory network. When put under estrogenic conditions PR gets activated and leads to a proliferative arrest by changing the ERα binding pattern to chromatin.
Publication of Detailed RIME Protocols
A detailed RIME protocol was published in 2016 in the journal Nature Protocols, again by lead researcher Hisham Mohammed at the University of Cambridge.
This article outlines the RIME protocol and covers the most important steps in the workflow. This article additionally discusses some ways to troubleshoot RIME experiments, and well as offering guidance regarding ways to optimize the protocol for different sample types.
The Next Generation: Multiplex RIME Techniques
Finally, the Carroll lab and collaborators set out to adapt RIME for multiplex analysis, which was described in a paper published in 2018 in the journal Nature Communications. In this new approach, which they call quantitative multiPLEXed Rapid Immunoprecipitation Mass spectrometry of Endogenous proteins (or qPLEX-RIME, for short), they combined the RIME method with chemical isobaric labeling using tandem mass tags (TMTs) to make the assay multiplexable.
For qPLEX-RIME, the initial RIME procedure was modified in the fixation step, where they used a double fixation using disuccinimidyl glutarate (DSG) and formaldehyde which has been previously applied in combination with ChIP assays to capture transient interactions more efficiently. Furthermore, they developed a downstream bioinformatics workflow (qPLEXanalyzer) which includes data processing, visualization, normalization, and statistical analysis.
With qPLEX-RIME it is possible to discriminate binding partners of the Protein of Interest from contaminant proteins and to characterize the dynamics of chromatin-associated protein complexes with higher sensitivity, reproducibility and increased throughput. The downstream analysis also enables fine-tuning of those parameters to fit the needs of the individual biological question.
How Does the RIME Protocol Work to Detect Protein-Protein Interactions?
As mentioned above, the main advantage of RIME is its sensitivity and the ability to target endogenous proteins with only low amounts of starting material. Furthermore, by using the same fixative and a similar sample preparation procedure, the proteomics data nicely complement the genomics data generated with ChIP.
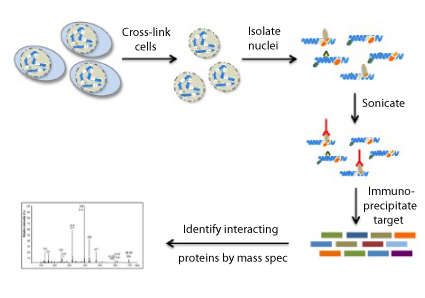
RIME Protocol Overview
The main steps of the RIME workflow are described below:
1. Immobilizing the Antibody on Beads
First, the beads need to be prepared for the Immunoprecipitation. For most RIME protocols, either protein A or G magnetic beads are used to immobilize the antibody of choice and washed to prepare them for the assay.
2. Crosslinking the Cell or Tissue Samples
After the antibody-bead complexes are prepared, samples can be processed for RIME. This step usually involved crosslinking the cells or tissues with formaldehyde, which is also used as a crosslinking agent in ChIP-Seq protocols. Crosslinking time and concentration of formaldehyde have been optimized from those ChIP-Seq protocols.
3. Preparation of Nuclear Lysates
In the next step of the RIME protocol, a nuclear lysate needs to be prepared. The cells are first lysed, and nuclei are separated, then nuclei are sonicated to prepare the chromatin for the immunoprecipitation reaction.
4. Immunoprecipitation
In this step, the antibody-bound beads and cell lysate are mixed and incubated overnight. After this incubation, the beads are washed to prepare them for the enzymatic digestion. We recommend performing the IP step in duplicate to generate more robust data. We also recommend performing control immunoprecipitations with non-specific normal IgG antibodies to control for background signal and non-specific association with antibodies or beads.
5. Enzymatic Digestion
Following the IP step, the proteins are pulled down are digested with trypsin to facilitate identification by mass spec. With RIME, samples are not alkylated or reduced before digestion to limit the number of IgG peptides, which improves the sensitivity of the assay.
6. Peptide Desalting by Solid-Phase Extraction (SPE)
The digested peptides now need to be prepared for LC-MS/MS analysis by performing a desalting step. This step is important because it reduces back-pressure on the HPLC system as a result of protein aggregation or high molecular weight species present in the digest supernatant. Protein binding is optimized by reloading the peptide flow-through up to three times.
7. Liquid Chromatograph with Tandem Mass Spectrometry (LC-MS/MS)
LC-MS/MS is an extremely effective method to identify unknown proteins. This technique combines liquid chromatography to separate peptides with the highly sensitive and specific mass analysis capability of tandem mass spectrometry. This step is usually performed by a core facility or collaborators with expertise in the mass spec field.
8. RIME Data Analysis
RIME data analysis generally includes evaluating the peptides identified in both of the IP reactions with the specific antibody but not the control IgG IPs. Gene ontology analysis is also often performed in RIME assays to identify biological pathways that the protein of interest participates in.
Recent RIME Publications & Discoveries
Since the initial RIME publication in 2013, many labs, especially those in the field of cancer research, have adopted the method in their research and already published the results. Further research and optimizations to the RIME protocol are being performed by our Epigenetic Services group and others.
Below, we summarize some publications that show the impact and applicability of the RIME technique.
RIME & Breast Cancer
A team of researchers lead by first author Robert Witwitcki from the Dana-Farber Cancer Institute in Boston characterized the transcription factor TRPS1 as a lineage-specific transcriptional repressor in breast cancer. They used RIME to characterize the mechanism by which TRPS1 represses transcription.
The team of researchers identified 214 and 147 TRSP1-associated proteins in HCC3153 and MDA-IBC3 breast cancer cells, respectively. When limiting the analysis to transcriptional and epigenetic regulators, they were able to find the NuRD complex and its components, including CHD4, MTA1-3, HDAC1/2, GATAD2B/A, as the top hits. These hits were validated with immunoblot assays and they also performed ChIP-Seq experiments to further validate their results.
Using RIME to Study Bromodomain Inhibitors
Enhancers are marked by the presence of certain epigenetic modifications, such as acetylation of histone H3 on lysine 27 (H3K27Ac), but how these modifications function at enhancers is not yet understood. A team from the Department of Discovery Oncology at Genentech started to investigate this epigenetic process.
Using the CBP/p300 bromodomain inhibitor GNE-049, the researchers were able to show that CBP/p300 specifically acetylates H3K27Ac. Furthermore, ChIP-Seq experiments indicated that CBP/p300 exclusively acts on enhancer regions. Using RIME, the scientists identified no change in the composition of p300 binding partners upon inhibitor treatment.
In summary, the team showed that CBP/p300 was crucial for the acetylation of H3K27 at enhancers and that this modification was required for sustaining enhancer activity. Their results also indicated that the bromodomain inhibitor they were studying did not interfere with the interactions between p300 and other protein co-factors.
Analyzing Chromatin-Remodeling Complexes with RIME
CHD1 is an ATP-dependent chromatin remodeler that localizes to promoters of actively-transcribed genes and facilitates transcriptional initiation through the eviction of nucleosomes.
This chromatin-remodeling protein is the one most commonly deleted in primary human prostate cancers, with an occurrence of approximately 15%. A team from Weill Cornell Medicine led by Michael Augello investigated the role of CHD1 in prostate cancer and published their results in the journal Cancer Cell.
First, they demonstrated, that CHD1 acts as a tumor suppressor in prostate cancer and that the tumor suppressor function likely is independent of its promoter-specific role. Then they used RIME to define the chromatin-bound interactome of CHD1 and compared it to a promoter-specific control using the H3K4me3 modification.
Their comparison revealed that 33% of the interactome was shared between factors identified bound to CHD1 and proteins associated promoters. They also found that the CHD1 interactome preferentially contained promoter-associated proteins. Further analysis showed that a large portion of the CHD1 interactome was unique to CHD1. A comparison with the interactome of the androgen receptor (AR) revealed that CHD1 and AR share 45% of their interactome, indicating a promoter-independent interactome of CHD1 and a potential role in its regulation.
Further experiments revealed that CHD1 can regulate nuclear receptor activity and indicated that CHD1 is a bona fide tumor suppressor within prostate tissue.
How to Get Started Using RIME in Your Research
To get RIME started in your lab you can consult us at Active Motif as we offer an end-to-end RIME service. To use our RIME service, you simply need to send us your samples (cells or tissue) and you’ll get your analyzed data back in a matter of weeks. We perform the chromatin preparation and sonication, the immunoprecipitation reactions in duplicate (including a negative control IP with normal IgG), prepare the samples for LC-MS/MS, perform the mass spectrometry, and deliver a final report that includes bioinformatic analysis.
The bioinformatics data analysis is performed by our in-house team of bioinformatics experts and results are summarized in an easy-to-digest project report. In addition to the raw data, the project report includes an “individual list”, which contains all identified proteins from each IP reaction after removing the background anti-IgG associated proteins, an additional “consolidated list”, which contains proteins common to both IPs after removing anti-IgG associated proteins, and a gene ontology report that groups the identified proteins into functional categories.
Setting up the RIME workflow in your lab is also an option, however, this might come down to the access to a mass spectrometer. If you do have access to such a device or service, you would need to find a good antibody that works in RIME. A good starting point is an antibody that works in ChIP, which then needs to be coupled to the beads. After optimizing and performing sample preparation and MS analysis, the bioinformatic analysis needs to be performed.
Although RIME offers the advantages of being able to study endogenous protein-protein interactions without the need to manipulate the sample on a genetic level, and the amount of starting material required is greatly reduced relative to historical methods, there are still some challenges to overcome when performing RIME. Therefore, it might be worthwhile to consider working with the epigenetics experts at Active Motif to generate high-quality RIME data as quickly as possible.
Summary: RIME is a Great Way to Identify Novel Protein-Protein Interactions on Chromatin and Discover New Protein Complexes
RIME represents a new method that enables researchers to identify the composition of chromatin-bound complexes from a low number of cells in a relatively brief workflow. As summarized above, this technique offers advantages over methods like IP-MS or ChIP-MS which rely on manipulating cells and overexpressing “tagged” proteins for affinity purification, which may lead to a number of disadvantages.
In contrast, the RIME assay purifies endogenous proteins without the need to genetically manipulate the cells. Furthermore, the RIME protocol allows for stringent washing conditions to reduce non-specific background proteins, thereby improving specificity and sensitivity.
RIME is also ideally suited to complement the genomics results obtained in ChIP-Seq studies with proteomics data because the same fixation and sample prep protocols are followed in both RIME and ChIP-Seq.
Contact us if you are interested in learning more about RIME and how our service works, or to request a quote.
About the author

Stefan Dillinger, Ph.D.
Stefan was born in the Free State of Bavaria, Germany. After studying biochemistry in Ulm and Regensburg, he got his Ph.D. in the field of epigenetics, studying the distribution of heterochromatin around nucleoli during cellular senescence. As a graduate student he started his own German science podcast “The Random Scientist” and is now the host of Active Motif’s Epigenetics Podcast. When Stefan is not working at Active Motif or recording podcasts, he is a passionate runner (he finished the New York City Marathon in 3 hours 21 minutes!!) and loves to spend time with his wife and son.
Contact Stefan on LinkedIn with any questions, or to get running advice.
Related Articles
Comprehensive Guide to Understanding and Using CUT&Tag Assays

March 9, 2020
CUT&Tag shows a lot of promise and has the potential to alleviate some ChIP limitations, but it also has its own set of limitations that must be considered. This article covers what CUT&Tag is and describes the advantages and drawbacks of this method.
Read More
Complete Guide to Using Reduced Representation Bisulfite Sequencing (RRBS) for Genome-Wide DNA Methylation Analysis

June 14, 2019
The RRBS method was first published in 2005, in the early days of NGS when sequencing was still expensive, as a way to get genome-wide single-nucleotide DNA methylation analysis for lower cost that whole genome bisulfite sequencing. This article covers what RRBS is and why it’s still a powerful method to study 5-mC.
Read More
<< Back to MOTIFvations Blog Home Page



