YourSeq (Full Transcript & 3’DGE) RNA-Seq Services Overview
This service is not available at this time.
Active Motif’s YourSeq (FT & 3’DGE) RNA-Seq Services provide a novel, efficient method for either conventional Full-Transcript (FT) or 3’-Digital Gene Expression (DGE) sequencing. Conventional full-transcript (FT) RNA-seq yields sequencing reads that map to the entire expressed transcriptome. The 3’ Digital Gene Expression (3’-DGE) option creates reads mapping just at the 3’-end of the transcript. Because of the reduced complexity, 3’-DGE requires a quarter to a third less sequencing depth, greatly reducing costs.
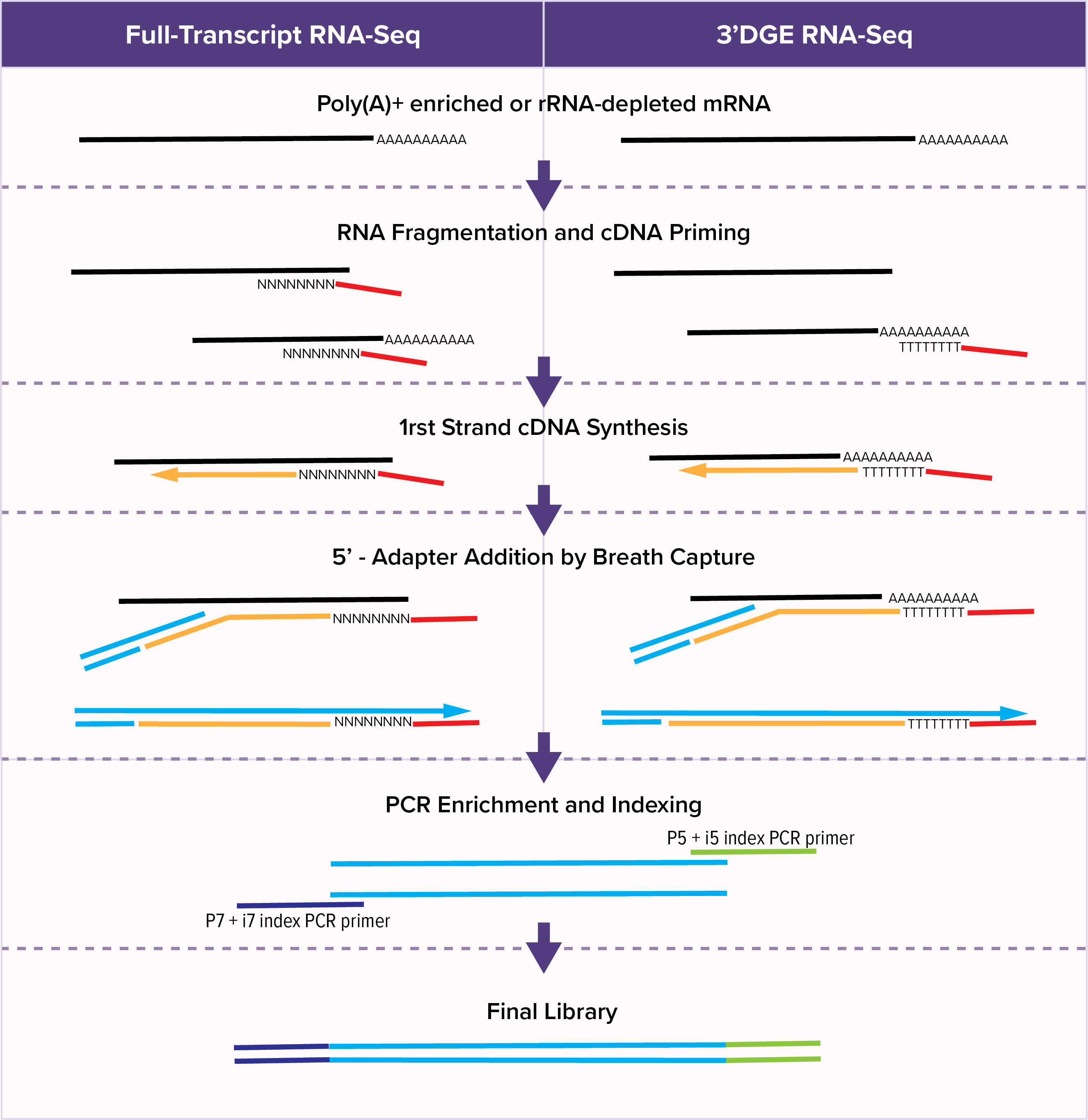
The YourSeq Library Prep workflow takes advantage of the natural phenomenon of DNA breathing to eliminate several steps which add time and cost to traditional RNA library prep protocols. DNA breathing is the opening and closing of DNA due to thermal fluctuations. Following first-strand cDNA synthesis, the YourSeq method adds the 5-prime adapter to the cDNA when the terminal end of the DNA:RNA hybrid molecule is open in a process called “Breath Capture.” This allows the enzymatic synthesis of a second strand of DNA without a step requiring the removal of RNA. This workflow also implements strictly enforced strand specificity, since the adapter is added by priming off the cDNA strand and cannot prime off the RNA strand.
YourSeq (FT & 3’DGE) RNA-Seq Services Highlights:
- Novel, improved method for sequencing mRNA allows for reduced sequencing depth and cost
- Option for both full-transcript (FT) or 3’-Digital Gene Expression (DGE) sequencing
- Strictly enforced strand specificity
(Click image to enlarge)
FT vs. 3’DGE
Active Motif offers two different YourSeq RNA-Seq Services: full-transcript (FT) or 3’-Digital Gene Expression (3’DGE). There are advantages and disadvantages to both methods. Use the appropriate workflow for your needs.
3’ Digital Gene Expression (3’DGE) Sequencing
The 3’ Digital Gene Expression (3’-DGE) option is best for differential gene expression analyses, where the main goal is to identify genes and pathways that are up- or down- regulated in response to some experimental condition.
Rather than random priming, the cDNA synthesis step uses an adapter with an oligo(dT) to prime off the transcript’s poly(A) tail. This means that 3’-DGE reads map to the 3’-end of the transcript. Since there is a theoretical 1-to-1 correlation between transcript molecule and cDNA molecule, transcript length is irrelevant and the number of reads mapping to a gene is solely a function of transcript abundance. A long gene, therefore, will have the same number of reads mapping to it as will a short gene of similar transcript abundance.
The reduced complexity associated with 3’-DGE confers one of the biggest advantages. Transcript counting means that less sequencing depth is needed for a robust analysis compared to conventional RNA-seq. Also, paired-end sequencing does not add value for 3’-DGE. So, where conventional RNA-seq libraries require 20-30 million pairs of paired-end reads per sample 3'DGE libraries would only require 8 million single reads at greatly reduced cost.
3’DGE RNA sequencing is optimal for:
- Differential gene expression for well-annotated genomes
- Improving annotation by identifying expressed genes and 3’-UTRs
- Genotyping and detection of polymorphisms in the 3’-UTR
- Detection and characterization of alternative polyadenylation
Full Transcript (FT) Sequencing
The Full Transcript (FT) Sequencing option is for conventional full transcript RNA sequencing. Conventional full-transcript (FT) coverage RNA-Seq yields sequencing reads that map to the entire expressed transcriptome.
This is achieved through the combination of random RNA fragmentation and subsequent random priming during the cDNA synthesis step. The result is sequencing reads that, when mapped to the reference transcriptome or genome, tile across the exons of expressed transcripts. With conventional RNA-seq, the number of reads mapping to a gene is a function of transcript abundance and transcript length. A long gene, therefore, will have more reads mapping to it than a short gene of similar transcript abundance.
FT sequencing requires 30 million paired-end reads per sample, while 3’-DGE libraries require only 8 million single reads. However, there are circumstances where full-transcript sequencing is required. For example, splice variant analysis, detection of exonic polymorphisms and the detection of gene fusion abundance would all require reads from the entire coding sequence.
Full-transcript RNA sequencing is optimal for:
- Differential gene expression for non-annotated genomes
- Genotyping and detection of exonic polymorphisms
- Splice variant analysis
- Detection of gene fusions abundance
YourSeq Services Data
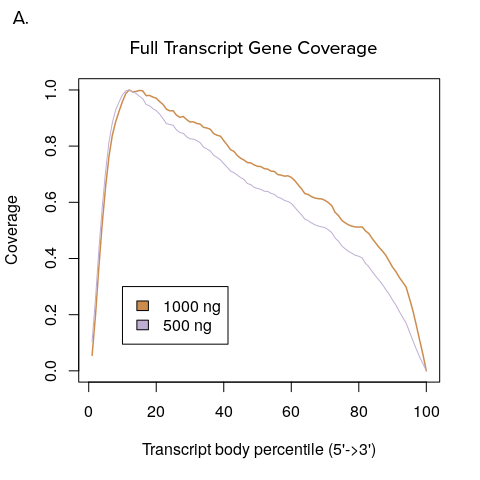
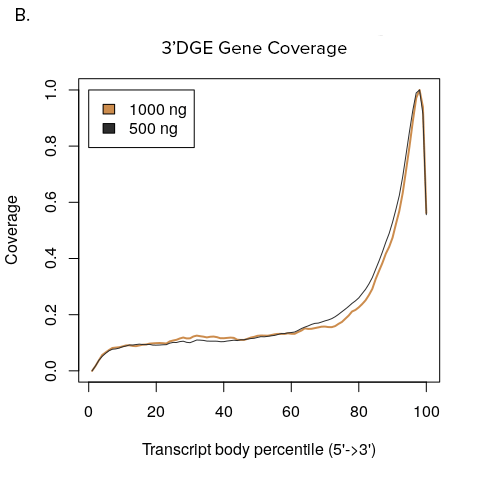
Figure 1: Transcript coverage for full transcript and 3’DGE sequencing.
A. YourSeq Full transcript sequencing reads tile across all exons of expressed transcripts. B. YourSeq 3’DGE sequencing reads map to the 3’-end of the transcript.
(Click image to enlarge)
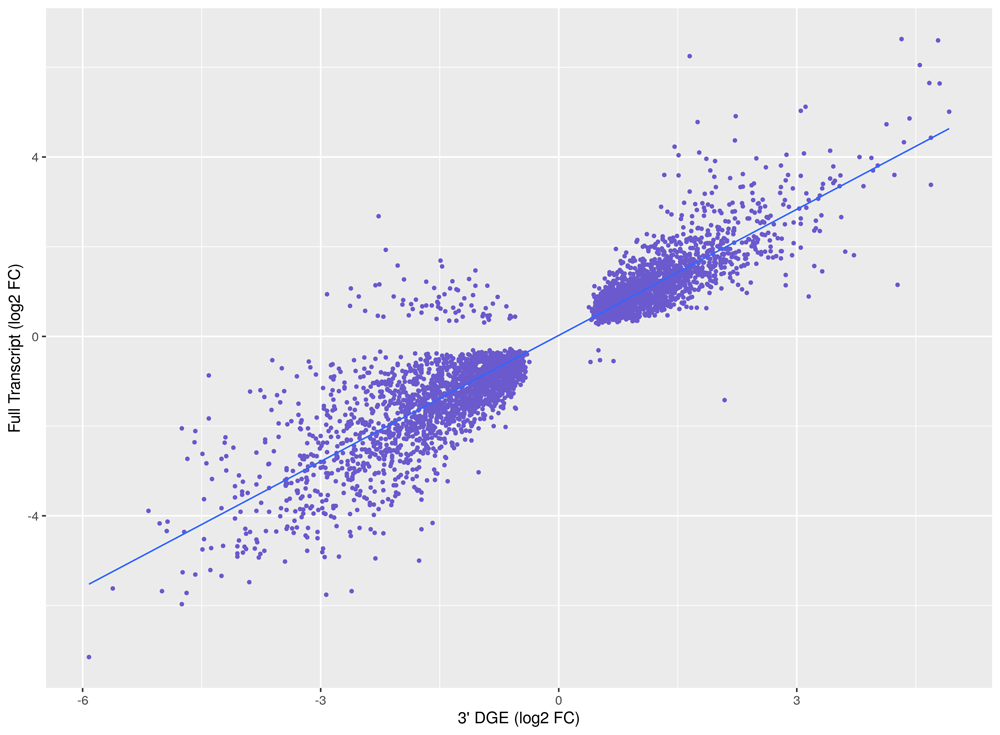
Figure 2: Correlation of gene counts for full transcript and 3’DGE sequencing.
A Log-log scatter plot of gene counts for full transcript versus 3’DGE shows similar gene capture, despite the lower sequencing reads required for 3’DGE. 1000 ng of total RNA was isolated from human leukemic cells (UT-7) and poly(A) enriched for removal of rRNA. The YourSeq (FT & 3’DGE) Strand-Specific mRNA Library Prep Kit was used to prepare libraries using either the full transcript or 3’DGE workflow. Libraries were pooled and sequenced on a NovaSeq 6000. Full transcript libraries were sequenced at 50 million paired-end reads and 3’DGE libraries were sequenced at 20 million single-end reads.
YourSeq Services Documents
YourSeq Services Sample Submission Portal
Our online sample submission portal allows you to easily upload your service project samples and track your project status. Follow the sample submission instructions in the portal to ensure that all your samples arrive at Active Motif in the best possible condition and properly associated with your project.